抗BCMA嵌合抗原受体、编码基因、重组表达载体及其构建方法和应用与流程
本发明属于肿瘤免疫治疗
技术领域:
,具体涉及一种抗BCMA嵌合抗原受体、编码基因、重组表达载体(尤其是一种基于复制缺陷性重组慢病毒的CAR-T转基因载体)及其构建方法和应用。
背景技术:
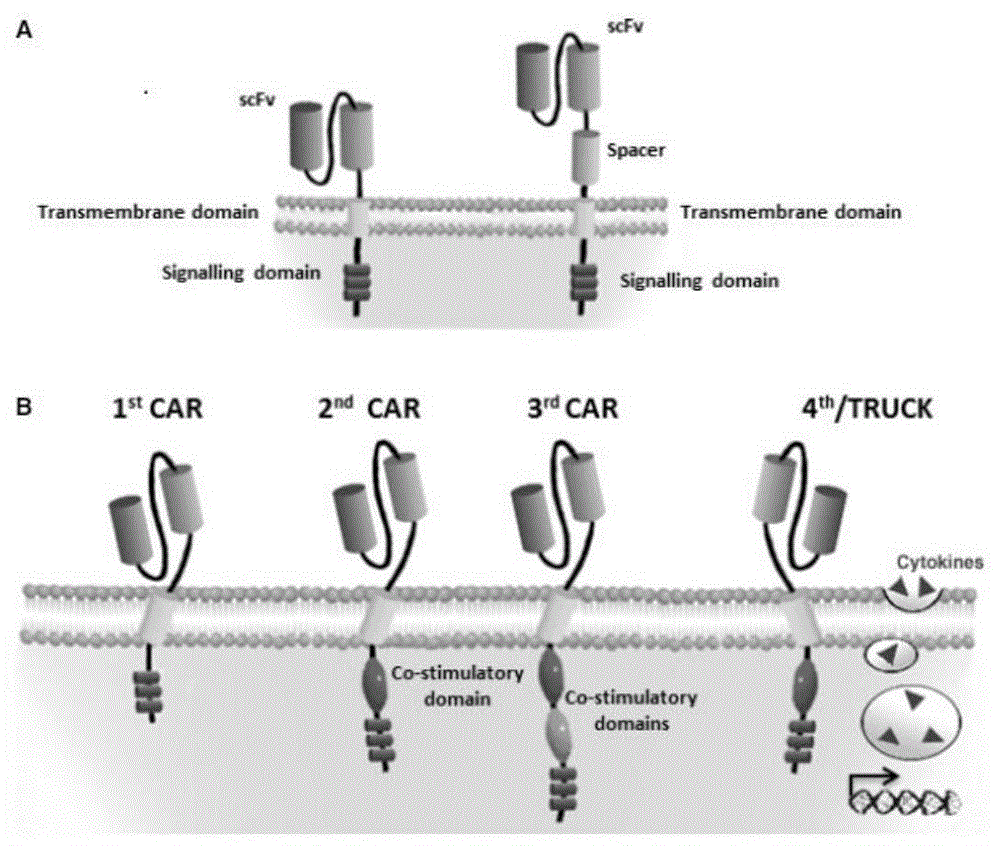
:肿瘤免疫治疗的理论基础是免疫系统具有识别肿瘤相关抗原、调控机体攻击肿瘤细胞(高度特异性的细胞溶解)的能力。这个生物过程十分复杂,目前仍处于研究之中。上世纪90年代,多个科研小组已经发现了肿瘤抗原(tμmorantigens),T淋巴细胞可以通过主要组织相容性复合体(majorhistocompatibilitycomplex,MHC)依赖性方式识别这些肿瘤抗原。肿瘤免疫治疗通常分为两类,非特异性免疫和特异性免疫。非特异性免疫治疗主要包括白细胞介素-2(interleμkin-2,IL-2),干扰素α(interferonα,IFN-α),肿瘤坏死因子(tμmornecrosisfactor,TNF-α),卡介苗等细胞因子和毒素,过继性细胞免疫治疗等。特异性免疫治疗主要是肿瘤疫苗。1.1肿瘤非特异性免疫治疗非特异性免疫应答是与生俱来的,它的形成并不需要抗原刺激,能广泛地针对多种抗原,是免疫应答的基础,但特异性不强,对某种特定抗原物质往往不能产生足够强度的反应非特异性的免疫治疗。在进入临床试验的多种细胞因子中,白细胞介素-2和干扰素应用最为广泛[RosenbergSA,LotzeMT,MμμlLM,etal.Aprogressreportonthetreatmentof157patientswithadvancedcancerμsinglymphokine-activatedkillercellsandinterleμkin-2orhigh-doseinterleμkin-2alone[J].NEnglJMed,1987,316(15):889-897.]。1.2肿瘤单克隆抗体的免疫治疗近20多年来单克隆抗体已在肿瘤治疗领域得到广泛应用。抗肿瘤单抗药物一般包括两类:一是抗肿瘤单抗,二是抗肿瘤单抗偶联物,或称免疫偶联物。免疫偶联物分子由单抗与“弹头”药物两部分组成,“弹头”主要包括放射性核素、药物和毒素,与单抗连接后分别构成放射免疫偶联物、化学免疫偶联物和免疫毒素。在1997年11月和1998年10月美国FDA分别通过了用于临床肿瘤治疗的两个单抗—Ritμximab(ritμxan)和Trastμzμmab(herceptin)[DillmanRO.Magicbμlletsatlast!Finally—approvalofamonoclonalantibodyforthetreatmentofcancer[J].CancerBiotherRadiopharm,1997,12:223-225.]。目前认为单抗的作用机制有阻断作用、信号传导作用以及靶向作用等三种作用机制,没有直接的杀伤作用。另外还存在药理学方面的问题,主要是到达肿瘤的药量不足。由于偶联物是异体蛋白,会被网状内皮系统摄取,有相当数量将积聚于肝、脾和骨髓。偶联物是大分子物质,通过毛细血管内皮层及穿透肿瘤细胞外间隙均受到限制。1.3肿瘤的过继免疫治疗肿瘤的过继免疫治疗是指将体外激活的自体或异体免疫效应细胞输注给患者,以杀伤患者体内的肿瘤细胞。肿瘤过继性免疫治疗中的一个关键问题是寻找合适的肿瘤杀伤细胞。自上世纪80年代以来,包括LAK、细胞因诱导的杀伤细胞(cytokine-indμcedkillers,CIK)、TIL等细胞已先后应用于临床,但由于存在着扩增倍速较低、细胞来源困难、细胞毒力不高等诸多问题,在临床应用上受到限制。如何提高T细胞的肿瘤抗原特异性具有重要的临床意义。T细胞对肿瘤抗原的识别主要是通过T细胞受体(Tcellreceptor,TCR)识别肿瘤细胞表面的人类白细胞抗原(hμmanleμkocyteantigen,HLA)-肽复合物,因此,T细胞对肿瘤抗原识别的特异性取决于T细胞表面的TCR。利用分子生物学的手段克隆肿瘤特异性T细胞的TCR,并通过构建含TCR的病毒载体,把TCR转入正常的T细胞中,使这些T细胞因携带肿瘤特异性而成为特异性肿瘤杀伤细胞[JohnsonLA,MorganRA,DμdleyME,etal.GenetherapywithhμmanandmoμseT-cellreceptorsmediatescancerregressionandtargetsnormaltissμesexpressingcognateantigen[J].Blood,2009,114(3):535-546.]。1.4肿瘤疫苗治疗肿瘤疫苗治疗是通过给患者体内导入肿瘤抗原来激发患者的特异性抗肿瘤免疫反应。由于疫苗治疗具有特异性、在体内免疫效应维持时间长等优点,目前已成为研究热点。近年来多肽疫苗、核酸疫苗、全蛋白疫苗、抗独特性抗体疫苗、重组病毒疫苗、细菌疫苗、基因修饰的肿瘤细胞疫苗、树突状细胞(dendriticcells,DC)疫苗等得到广泛研究和应用[RobbinsPF,MorganRA,FeldmanSA,etal.TμmorregressioninpatientswithmetastaticsynovialcellsarcomaandmelanomaμsinggeneticallyengineeredlymphocytesreactivewithNY-ESO-1[J].JClinOncol,2011,29(7):917-924.]。肿瘤疫苗治疗的大规模应用还有三个方面的问题亟待解决。首先,肿瘤相关抗原,每个肿瘤、每个亚型、每个肿瘤分期,这些相对抗原表达是不一样的,所以选准抗原,选准病人人群是致关重要的。第二,如何达到肿瘤抗原在树突状细胞中髙效吸收与表达?抗原被树突状细胞吸收是以表面受体为介导的。树突状细胞有十几个受体,如何根据特定的抗原选择相应的受体?第三,针对树突状细胞分化成熟的调控。树突状细胞的分化成熟是一个非常复杂的过程,它既可走向激活T细胞也可以走向抑制T细胞。[靶向DC细胞的治疗型肿瘤疫苗:亮点与挑战并存。http://www.biodiscover.com/news/research/115794.html]1.5肿瘤CAR-T治疗CAR-T,全称是ChimericAntigenReceptorT-CellImmμnotherapy,嵌合抗原受体T细胞免疫疗法,结构如图1所示[EleanorJ.Cheadle,etal.CARTcells:drivingtheroadfromthelaboratorytotheclinic.ImmμnologicalReviews2014.Vol.257:91–106]。第一代CAR介导的T细胞激活是通过CD3z链或FceRIg上的酪氨酸激活基序完成的。CD3z链能够提供T细胞激活、裂解靶细胞、调节IL-2分泌以及体内发挥抗肿瘤活性所需的信号。但第一代CAR改造T细胞的抗肿瘤活性在体内受到了限制,T细胞增殖减少最终导致T细胞的凋亡。第二代CAR在胞内增加了一个新的共刺激信号,实验证明,这使得原有的使源自TCR/CD3复合体的“信号1”扩大,许多研究都表明,搭载了“信号2”的第二代CAR与第一代CAR相比,抗原特异性不变,T细胞增殖、细胞因子分泌增加,抗细胞凋亡蛋白分泌增加,细胞死亡延迟。常用的共刺激分子为CD28,但之后有研究将CD28用CD137(4-1BB)进行替换,除此之外,一种使用NK细胞受体CD244的思路也被提出来。虽然不同的第二代CAR究竟孰优孰劣,不同的研究者用不同的肿瘤在体内和体外的研究中得到的结果不尽相同。[深度完整版:CAR-T的现状和未来.生物谷.2015-051-15]为了进一步改良CAR的设计,许多研究组开始着眼于发展第三代CAR,不仅包括“信号1”、“信号2”,还包括了额外的共刺激信号。不同研究者们用不同的靶点和共刺激信号开展的研究所得到的第二代CAR和第三代CAR的比较结果存在一定的差异性。一些研究报道表达第三代CAR的重组T细胞在抗肿瘤活性、存活周期及细胞因子释放方面均显著提高;Wilkie等的研究结果显示靶向MΜC1的第二代CAR与第三代CAR重组T细胞在抗肿瘤细胞毒性方面并无明显差异,虽然表达第三代CAR的T细胞能够分泌更大量的IFN-γ(WilkieS,PiccoG,FosterJ,etal.RetargetingofhμmanTcellstotμmorassociatedMΜC1:theevolμtionofachimericantigenreceptor.JImmμnol2008;180:4901–4909.)。值得注意的是,上述区别仅仅是体外实验中获得的结论,目前尚未在体内比较第二代和第三代CAR的报道。这几代CAR之间的差异可能不止来自于信号传导域,胞外的抗原结合域(scFv)、重组T细胞的转染方法(慢病毒VS逆转录病毒)、重组T细胞的回输方式(静脉回输VS腹膜VS瘤体)等均可能影响CAR-T细胞的最终抗肿瘤效果。技术实现要素:本发明的目的在于克服现有技术的不足,提供了一种抗BCMA嵌合抗原受体、编码基因、重组表达载体及其构建方法和应用。本发明的目的通过以下技术方案来实现:本发明的第一目的在于提供一种抗BCMA嵌合抗原受体(二代CAR),包括依次串联的如SEQIDNO.15所示的CD8leader嵌合受体信号肽、如SEQIDNO.16所示的BCMA单链抗体重链VH、如SEQIDNO.19所示的OptimalLinkerC、如SEQIDNO.20所示的BCMA单链抗体轻链VL、如SEQIDNO.21所示的CD8Hinge嵌合受体铰链、如SEQIDNO.22所示的CD8Transmembrane嵌合受体跨膜区、如SEQIDNO.23所示的CD137嵌合受体共刺激因子,以及如SEQIDNO.24所示的TCR嵌合受体T细胞激活域。进一步的,所述嵌合抗原受体的氨基酸序列如SEQIDNO.50所示。一种抗BCMA嵌合抗原受体(三代CAR),包括依次串联的如SEQIDNO.15所示的CD8leader嵌合受体信号肽、如SEQIDNO.16所示的BCMA单链抗体重链VH、如SEQIDNO.19所示的OptimalLinkerC、如SEQIDNO.20所示的BCMA单链抗体轻链VL、如SEQIDNO.21所示的CD8Hinge嵌合受体铰链、如SEQIDNO.22所示的CD8Transmembrane嵌合受体跨膜区、如SEQIDNO.25所示的CD28嵌合受体共刺激因子、如SEQIDNO.23所示的CD137嵌合受体共刺激因子、以及如SEQIDNO.24所示的TCR嵌合受体T细胞激活域。进一步的,所述嵌合抗原受体的氨基酸序列如SEQIDNO.51所示。本发明的第二目的在于提供一种编码上述抗BCMA嵌合抗原受体的基因。进一步的,所述基因的核苷酸序列如SEQIDNO.52(二代CAR)或SEQIDNO.53(三代CAR)所示。本发明的第三目的在于提供含有上述基因的重组表达载体。进一步的,所述重组表达载体为慢病毒表达载体、逆转录病毒表达载体、腺病毒表达载体、腺相关病毒表达载体或质粒。进一步的,所述的慢病毒表达载体包含上述抗BCMA嵌合抗原受体的基因。进一步的,所述的慢病毒表达载体包括:用于质粒复制的原核复制子pUCOri序列,如SEQIDNO.2所示;用于目的菌株大量扩增的含氨苄青霉素抗性基因AmpR序列,如SEQIDNO.1所示;用于增强真核细胞内的复制的病毒复制子SV40Ori序列,如SEQIDNO.3所示;用于慢病毒包装的慢病毒包装顺式元件;用于真核细胞表达绿色荧光的ZsGreen1绿色荧光蛋白,如SEQIDNO.11所示;用于共同转录表达蛋白质的IRES核糖体结合序列,如SEQIDNO.12所示;用于嵌合抗原受体基因的真核转录的人EF1α启动子,如SEQIDNO.14所示;用于组成集识别、传递、启动于一体的二代CAR或三代CAR的嵌合抗原受体的编码基因,如SEQIDNO.52或SEQIDNO.53所示;用于增强转基因的表达效率的eWPRE增强型土拨鼠乙肝病毒转录后调控元件,如SEQIDNO.13所示。进一步的,所述慢病毒包装顺式元件采用第二代慢病毒载体包括:如SEQIDNO.5所示的慢病毒5terminalLTR、如SEQIDNO.6所示的慢病毒3terminalSelf-InactivatingLTR、如SEQIDNO.7所示的Gag顺式元件、如SEQIDNO.8所示的RRE顺式元件、如SEQIDNO.9所示的env顺式元件、如SEQIDNO.10所示的cPPT顺式元件。进一步的,所述慢病毒包装顺式元件采用第三代慢病毒载体包括:如SEQIDNO.5所示的慢病毒5terminalLTR、如SEQIDNO.6所示的慢病毒3terminalSelf-InactivatingLTR、如SEQIDNO.7所示的Gag顺式元件、如SEQIDNO.8所示的RRE顺式元件、如SEQIDNO.9所示的env顺式元件、如SEQIDNO.10所示的cPPT顺式元件所述慢病毒包装顺式元件,以及如SEQIDNO.4所示的RSV启动子。进一步的,所述eWPRE增强型土拨鼠乙肝病毒转录后调控元件有6个核苷酸的增强突变,具体为:g.396G>A、g.397C>T、g.398T>C、g.399G>A、g.400A>T、g.411A>T。本发明的第四目的在于提供一种上述慢病毒表达载体的构建方法,包括以下步骤:(1)将含氨苄青霉素抗性基因AmpR序列(如SEQIDNO.1所示)、原核复制子pUCOri序列(如SEQIDNO.2所示)、病毒复制子SV40Ori序列(如SEQIDNO.3所示)、用于慢病毒包装的慢病毒包装顺式元件、ZsGreen1绿色荧光蛋白(如SEQIDNO.11所示)、IRES核糖体结合序列(如SEQIDNO.12所示)、eWPRE增强型土拨鼠乙肝病毒转录后调控元件(如SEQIDNO.13所示)存储于慢病毒骨架质粒上;(2)将人EF1α启动子(如SEQIDNO.14所示)、用于组成集识别、传递、启动于一体的二代CAR或三代CAR的嵌合抗原受体组合成二代CAR或三代CAR设计方案,经过酶切、连接、重组反应克隆至慢病毒骨架质粒中,得到二代CAR或三代CAR设计的重组慢病毒质粒;(3)将得到的重组慢病毒质粒与慢病毒包装质粒pPac-GP、pPac-R以及膜蛋白质粒pEnv-G共同转染HEK293T/17细胞,在HEK293T/17细胞中进行基因转录表达后,包装成功重组慢病毒载体会释放到细胞培养上清中,收集包含的重组慢病毒载体的上清液;(4)将得到的重组慢病毒上清采用抽滤、吸附、洗脱的离子交换方式进行纯化,分别得到重组慢病毒载体。进一步的,步骤(1)中,所述慢病毒包装顺式元件采用第二代慢病毒载体包括:如SEQIDNO.5所示的慢病毒5terminalLTR、如SEQIDNO.6所示的慢病毒3terminalSelf-InactivatingLTR、如SEQIDNO.7所示的Gag顺式元件、如SEQIDNO.8所示的RRE顺式元件、如SEQIDNO.9所示的env顺式元件、如SEQIDNO.10所示的cPPT顺式元件;所述慢病毒包装顺式元件采用第三代慢病毒载体包括:如SEQIDNO.5所示的慢病毒5terminalLTR、如SEQIDNO.6所示的慢病毒3terminalSelf-InactivatingLTR、如SEQIDNO.7所示的Gag顺式元件、如SEQIDNO.8所示的RRE顺式元件、如SEQIDNO.9所示的env顺式元件、如SEQIDNO.10所示的cPPT顺式元件所述慢病毒包装顺式元件,以及如SEQIDNO.4所示的RSV启动子。进一步的,步骤(2)中,所述用于组成集识别、传递、启动于一体的二代CAR的嵌合抗原受体包括:如SEQIDNO.15所示的CD8leader嵌合受体信号肽、如SEQIDNO.16所示的BCMA单链抗体重链VH轻链VL、如SEQIDNO.19所示的OptimalLinkerC、如SEQIDNO.20所示的BCMA单链抗体轻链VL、如SEQIDNO.21所示的CD8Hinge嵌合受体铰链、如SEQIDNO.22所示的CD8Transmembrane嵌合受体跨膜区、如SEQIDNO.23所示的CD137嵌合受体共刺激因子、如SEQIDNO.24所示的TCR嵌合受体T细胞激活域;所述用于组成集识别、传递、启动于一体的三代CAR的嵌合抗原受体包括:如SEQIDNO.15所示的CD8leader嵌合受体信号肽、如SEQIDNO.16所示的BCMA单链抗体重链VH轻链VL、如SEQIDNO.19所示的OptimalLinkerC、如SEQIDNO.20所示的BCMA单链抗体轻链VL、如SEQIDNO.21所示的CD8Hinge嵌合受体铰链、如SEQIDNO.22所示的CD8Transmembrane嵌合受体跨膜区、如SEQIDNO.25所示的CD28嵌合受体共刺激因子、如SEQIDNO.23所示的CD137嵌合受体共刺激因子、以及如SEQIDNO.24所示的TCR嵌合受体T细胞激活域。进一步的,步骤(1)中,所述eWPRE增强型土拨鼠乙肝病毒转录后调控元件有6个核苷酸的增强突变,具体为:g.396G>A、g.397C>T、g.398T>C、g.399G>A、g.400A>T、g.411A>T。进一步的,步骤(2)中,由人EF1α启动子启动整个CAR基因表达;CD8leader嵌合受体信号肽位于CAR编码序列的N端,用于引导CAR蛋白定位于细胞膜;BCMA单链抗体重链VH、OptimalLinkerC、BCMA单链抗体轻链VL组合成scfv区域,用于识别BCMA抗原;CD8Hinge嵌合受体铰链用于将scfv锚定于细胞膜外侧;CD8Transmembrane嵌合受体跨膜区用于将整个嵌合受体固定于细胞膜上;CD137嵌合受体共刺激因子用于刺激T细胞增殖和细胞因子分泌;TCR嵌合受体T细胞激活域用于激活下游信号通路的表达;当scfv区域与BCMA抗原结合时,信号通过嵌合受体传递至细胞内,从而产生包括T细胞增殖、细胞因子分泌增加、抗细胞凋亡蛋白分泌增加、细胞死亡延迟、裂解靶细胞的一系列生物学效应。进一步的,步骤(4)中,所述慢病毒载体有带荧光标签zsGreen1的版本和不带荧光标签zsGreen1版本,带荧光标签的版本用于体外实验,不带荧光标签的版本用于临床实验。进一步的,步骤(4)中,所述抽滤步骤控制上清体积在200ml~2000ml,真空度控制在-0.5MPA~-0.9MPA,防止由于堵孔带来的载体损失;所述吸附步骤控制溶液的PH值在6~8,防止PH的变化导致载体失活;所述洗脱步骤控制洗脱液的离子强度在0.5M~1.0M,防止离子强度的变化导致洗脱不完全或者载体失活。本发明所采用的表达载体包括原核复制子(pUCori)用于质粒复制;原核筛选标记(AmpR)用于目的菌株大量扩增;病毒复制子(SV40Ori)用于增强真核细胞内的复制;慢病毒包装顺式元件(RSV、5terminalLTR、3terminalSelf-InactivatingLTR、Gag、RRE、env、cPPT)用于慢病毒包装;真核荧光标签蛋白(ZsGreen1)用于真核细胞表达绿色荧光;共表达元件(IRES)用于共同转录表达蛋白质;真核启动子(EF1α)用于嵌合抗原受体基因的真核转录;嵌合抗原受体(CD8leader、BCMAVH、CommonLinkerA(SEQIDNO.17)/CommonLinkerB(SEQIDNO.18)/OptimalLinkerC(SEQIDNO.19)、BCMAVL、CD8Hinge、CD8Transmembrane、CD137、TCR)用于组成集识别、传递、启动于一体的二代和三代CAR;转录后调控元件(eWPRE)用于增强转基因的表达效率。本发明的第五目的在于提供一种CAR-T细胞,所述的CAR-T细胞是由上述抗BCMA嵌合抗原受体修饰的T淋巴细胞。本发明的再一目的在于提供上述CAR-T细胞在制备多发性骨髓瘤治疗药物中的应用。本发明涉及含肽的医药配置品,具体涉及:一、含氨苄青霉素抗性基因AmpR序列、原核复制子pΜCOri序列,、病毒复制子SV40Ori序列、RSV启动子、人EF1α启动子、慢病毒5terminalLTR、慢病毒3terminalSelf-InactivatingLTR、Gag顺式元件、RRE顺式元件、env顺式元件、cPPT顺式元件、IRES核糖体结合序列、ZsGreen1绿色荧光蛋白、WPRE土拨鼠乙肝病毒转录后调控元件的重组慢病毒载体骨架,这种重组慢病毒载体骨架可以搭载不同的治疗性基因并广泛的用于过继性细胞治疗领域。二、重组慢病毒载体骨架、CD8leader嵌合受体信号肽、BCMA单链抗体重链VH、单链抗体铰链LinkerA、单链抗体铰链LinkerB、单链抗体铰链LinkerC、BCMA单链抗体轻链VL、CD8Hinge嵌合受体铰链、CD8Transmembrane嵌合受体跨膜区、CD28嵌合受体共刺激因子、CD137嵌合受体共刺激因子、TCR嵌合受体T细胞激活域构建形成重组慢病毒载体,该方法得到的重组慢病毒载体可以实现在人T淋巴细胞上表达BCMA嵌合抗原受体,引导并激活T淋巴细胞对BCMA阳性细胞的杀伤作用,在临床上用于治疗多发性骨髓瘤。本发明所采用的针对BCMA的CAR-T技术,是一种综合了肿瘤单克隆抗体的免疫治疗和肿瘤的过继免疫治疗优点的靶向治疗新技术。多发性骨髓瘤(Multiplemyeloma)是一种通常很难治愈的恶性浆细胞肿瘤,而BCMA(B-cellmaturationantigen)作为一种标志物选择性表达在B细胞(包括多发性骨髓瘤细胞)表面,可以作为CAR-T细胞的靶标引导CAR-T细胞杀伤肿瘤细胞(B-cellMaturationAntigenisaPromisingTargetforAdoptiveTcellTherapyofMultipleMyeloma.RobertO.Carpenter,etal.ClinCancerRes.2013April15;19(8):2048–2060.)。最近,在上www.clinicaltrials.gov有两项针对BCMA的多发性骨髓瘤CAR-T治疗的项目正在招募患者,注册号分别是#NCT02546167和#NCT02215967,CAR-T治疗被认为是最有前景的多发性骨髓瘤治疗方式之一。嵌合抗原受体(CAR)是CAR-T的核心部件(图1所示),赋予T淋巴细胞HLA非依赖的方式识别肿瘤抗原的能力,这使得经过CAR改造的T细胞相较于天然T细胞表面受体TCR能够识别更广泛的目标。CAR的基础设计中包括一个肿瘤相关抗原(tumor-associatedantigen,TAA)结合区(通常来源于单克隆抗体抗原结合区域的scFV段),一个胞外铰链区,一个跨膜区和一个胞内信号转导区。scFV段的设计对于CAR的特异性、有效性以及基因改造T细胞自身的安全性来是关键的决定因素。与现有技术相比,本发明具有如下有益效果:本发明采用的scFV段的linker设计,是使用世翱公司的Linkerspool,经过蛋白质结构生物信息学数据库(https://www.predictprotein.org/)的分析,通过对蛋白质二级结构、溶剂可接触性、蛋白质柔韧性、二硫键桥、结合位点等蛋白质特性的比较,优选得出。通过体外细胞因子分泌检测试验以及杀伤效率试验证明,与国外的设计相比,能够显著提高CAR-T细胞的体外杀伤作用。并且,在临床治疗的效果上也比国外临床实验的效果好。该scFV段的linker设计,同样可以应用于第三代CAR设计方案。第三代CAR设计与第二代设计相比,增加了CD28嵌合受体共刺激因子(SEQIDNO.25),会有更强的信号放大作用。本发明所采用的慢病毒载体柱纯化系统,系本申请人开发出的慢病毒规模化生产工艺。常用的超速离心法或者高速离心法,是利用离心沉降原理分离慢病毒颗粒,不可避免的会残留很多沉降系数相近的杂质,对后续实验带来不利影响。并且,装管过程复杂、操作繁琐、多次转换容器带来更多的污染机会。而本申请人开发的慢病毒载体柱纯化工艺为半自动化操作,全部过程在百级实验区域完成,避免人工操作的繁琐和污染几率,所回收的慢病毒载体在内毒素、支原体等指标上完全达到临床标准。后续可跟进开发全自动纯化仪。本发明所采用CAR设计方案也可以应用于第二代慢病毒载体结构上。第二代和第三代慢病毒载体在结构上的区别(如图2B所示),主要是第三代慢病毒载体把第二代载体5’LTR的U3区域替换为RSV启动子,这样就消除了U3转录时对Tat蛋白的依赖,既可以在慢病毒的结构基因里去除Tat序列,也提高了慢病毒基因组转录水平和转录持续性。第二代和第三代慢病毒载体主要是基因组转录方式的区别,因此本发明所采用CAR设计方案可以应用于这两代慢病毒载体。本发明采用的第三代慢病毒骨架质粒pLenti-3Gbasic,与宾州大学CarlH.June等人(PorterDL,LevineBL,KalosM,BaggA,JuneCH.ChimericantigenreceptormodifiedTcellsinchroniclymphoidleukemia.NEnglJMed2011;365:725-33.)所采用的第三代慢病毒载体相比,去除了噬菌体f1复制起点,采用真核病毒SV40复制子,增加了目的基因在真核细胞内的拷贝数,增强了真核表达效果。本发明采用的的第三代慢病毒骨架质粒pLenti-3Gbasic,采用enhancedWPRE元件,与宾州大学CarlH.June等人(PorterDL,LevineBL,KalosM,BaggA,JuneCH.ChimericantigenreceptormodifiedTcellsinchroniclymphoidleukemia.NEnglJMed2011;365:725-33.)所采用的WPRE元件相比,有6个核苷酸的增强突变(g.396G>A、g.397C>T、g.398T>C、g.399G>A、g.400A>T、g.411A>T),能够增强初级转录产物的多聚腺苷化,增加细胞内mRNA的含量,增强转基因的表达效率。本发明采用的Lentival包装系统是无辅助病毒的四质粒包装系统,通过四种质粒共同转染至HEK293T/17细胞中,产生重组慢病毒载体。重组后的慢病毒载体是复制缺陷型载体,能将外源片段整合入宿主基因,一次性使用,无法复制和增殖,安全性有很大提高。本发明采用的慢病毒载体有带荧光标签zsGreen1的版本和不带荧光标签zsGreen1版本,带荧光标签的版本用于体外实验,不带荧光标签的版本用于临床实验。本发明优选采用第三代慢病毒载体(图2A所示),3’SINLTR去除了U3区域,消除了慢病毒载体自我复制的可能性,大大提高了安全性;增加了cPPT和WPRE元件,提高了转导效率和转基因的表达效率;采用RSV启动子保证了慢病毒载体包装时核心RNA的持续高效转录;采用人自身的EF1α启动子,使CAR基因能够在人体内长时间持续表达。可见,本发明所述的重组慢病毒载体将给多发性骨髓瘤的CAR-T治疗提供可靠的转基因保障。附图说明图1是本发明所述的CAR的示意图,其中,图1(A)是CAR的基本结构图,图1(B)是CAR的代次改进示意图;图2是本发明所述的慢病毒载体结构示意图;其中,图2(A)是本发明采用的第三代慢病毒载体结构示意图,图2(B)是第二代和第三代慢病毒载体结构比较;图3是本发明实施例1中构建本发明所述的重组慢病毒载体的构建流程图,其中,图3(A)是慢病毒骨架质粒pLenti-3Gbasic的结构示意图;图3(B)是pCAR-BCMA-CLA质粒的结构示意图;图3(C)图是pCAR-BCMA-CLB质粒的结构示意图;图3(D)图是pCAR-BCMA-OLC质粒的结构示意图;图3(E)图是慢病毒包装质粒pPac-GP的结构示意图;图3(F)图是慢病毒包装质粒pPac-R的结构示意图;图3(G)图是膜蛋白pEnv-G的结构示意图;图4是本发明实施例1中重组慢病毒质粒pCAR138-CLA的酶切预测及酶切琼脂糖凝胶电泳图;其中,图4A是重组慢病毒质粒pCAR-BCMA-CLA的酶切预测示意图,其中,lane1是1kbDNAladderMarker:条带从上到下依次为:10kb、8Kb、6kb、5Kb、4kb、3.5Kb、3Kb、2.5kb、2Kb、1.5kb、1Kb、750bp、500bp、250bp;lane2是pCAR-BCMA-CLA的SalI酶切预测:条带从上到下依次为:8613bp、818bp、459bp;图4B是重组慢病毒质粒pCAR-BCMA-CLA的酶切琼脂糖凝胶电泳图;其中,lane1是1kbDNAladderMarker的电泳结果;lane2是pCAR-BCMA-CLA的SalI酶切电泳结果;图5是本发明实施例1中重组慢病毒质粒pCAR-BCMA-CLB的酶切预测及酶切琼脂糖凝胶电泳图;其中,图5A是重组慢病毒质粒pCAR-BCMA-CLB的酶切预测示意图,其中,lane1是1kbDNAladderMarker:条带从上到下依次为:10kb、8Kb、6kb、5Kb、4kb、3.5Kb、3Kb、2.5kb、2Kb、1.5kb、1Kb、750bp、500bp、250bp;lane2是pCAR-BCMA-CLB的PvuI酶切预测:条带从上到下依次为:5248bp、3497bp、896、249;图5B是重组慢病毒质粒pCAR-BCMA-CLB的酶切琼脂糖凝胶电泳图;其中,lane1是1kbDNAladderMarker的电泳结果;lane2是pCAR-BCMA-CLB的PvuI酶切电泳结果;图6是本发明实施例1中重组慢病毒质粒pCAR-BCMA-OLC的酶切预测及酶切琼脂糖凝胶电泳图;其中,图6A是重组慢病毒质粒pCAR-BCMA-OLC的酶切预测示意图,其中,lane1是1kbDNAladderMarker:条带从上到下依次为:10kb、8Kb、6kb、5Kb、4kb、3.5Kb、3Kb、2.5kb、2Kb、1.5kb、1Kb、750bp、500bp、250bp;lane2是pCAR-BCMA-OLC的KpnI酶切预测:条带从上到下依次为:8464bp、1426bp;图6B是重组慢病毒质粒pCAR-BCMA-OLC的酶切琼脂糖凝胶电泳图,其中,lane1是1kbDNAladderMarker的电泳结果;lane2是pCAR-BCMA-OLC的KpnI酶切电泳结果;图7是本发明实施例2中离子交换色谱法纯化重组慢病毒载体的流程图;图8是本发明实施例2中重组慢病毒载体的不同纯化方式的滴度检测结果示意图;图9是本发明实施例2中重组慢病毒载体的不同纯化方式的支原体检测结果示意图;其中,lane1为DL2000marker,从上到下条带条带从上到下依次为:2kb、1kb、750bp、500bp、250bp、100bp;lane2为阳性对照;lane3为阴性对照;lane4为PBS;lane5为水;lane6为裂解液;lane7为超速离心纯化的慢病毒;lane8为高速离心纯化的慢病毒;lane9为离子交换色谱纯化的慢病毒;lane10为空细胞;图10是本发明实施例3中mRNA相对表达量的柱状图,RT-QPCR结果表明CAR在PBMC细胞内高效转录;图11是本发明实施例3中CAR蛋白表达量的WB检测图,结果表明CAR蛋白在PBMC细胞内高效表达;其中,图11A中,lane1为PBMC空细胞,lane2为对照病毒MOCK,lane3为lvCAR-BCMA-CLA,lane4为lvCAR-BCMA-CLB,lane5为lvCAR-BCMA-OLC;图11B是beta-actin内参条带;图12是本发明实施例3中LDH检测不同效靶比条件下的杀伤效率,E为效应细胞,T为靶细胞;图13是本发明实施例3中qPCR检测不同效靶比条件下细胞因子表达水平示意图,E为效应细胞,T为靶细胞;其中,图13A表示IL-2的mRNA转录水平;图13B表示IFN-γ的mRNA转录水平;图14是本发明实施例3中CAR-BCMA-T细胞在体外和体内的变化情况;其中,图14A表示anti-bcma2-T细胞及其对照SP6-T细胞在体外对H929细胞的杀伤作用曲线;图14B表示anti-bcma2-T细胞及其对照SP6-T细胞在体外对RPMI8226细胞的杀伤作用曲线;图14C表示回输后,小鼠体内肿瘤体积随时间的变化情况;图14D表示回输后,小鼠生存期随时间的变化情况;图15是本发明实施例4中连续3个批次的第二代和第三代重组慢病毒载体的滴度结果比较;图16为本发明实施例5中LDH检测不同效靶比条件下的杀伤效率,其中,E为效应细胞,T为靶细胞。具体实施方式以下实施例仅用于说明本发明而不用于限制本发明的范围。实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。实施例1构建重组慢病毒载体一、材料1、慢病毒骨架质粒pLenti-3Gbasic,慢病毒包装质粒pPac-GP、pPac-R以及膜蛋白质粒pEnv-G,HEK293T/17细胞、同源重组酶由世翱(上海)生物医药科技有限公司提供;2、引物:根据引物设计原则设计扩增DNA片段和靶位点所需的引物,该引物由上海生物公司合成,具体为:EF1α-F:5’-ATTCAAAATTTTATCGATGCTCCGGTGCCCGTCAGT-3’(SEQIDNO.26)EF1α-R:5’-TCACGACACCTGAAATGGAAGA-3’(SEQIDNO.27)CD8leader-F:5’-GGTGTCGTGAGGATCCGCCACCATGGCCTTACCAGTGACCGC-3’(SEQIDNO.28)CD8leader-R:5’-GTGTCATCTGGATGTCCGGCCTGGCGGCGTG-3’(SEQIDNO.29)VH-F:5’-CACGCCGCCAGGCCGCAGATTCAGCTGGTGCAGAGC-3’(SEQIDNO.30)VH-R:5’-GCTGCTCACGGTCAGGGTG-3’(SEQIDNO.31)CLA-VL-F:5’-CTGACCGTGAGCAGCGGCGGTGGCTCGGGTGGTGGGTCGGGCGGCGGATCTGGGGGAGGTTCTGATATTGTGCTGACCCAGAGCC-3’(SEQIDNO.32)CLB-VL-F:5’-CTGACCGTGAGCAGCGGATCCACCTCCGGATCCGGAAAACCCGGATCCGGAGAAGGATCCACCAAAGGAGATATTGTGCTGACCCAGAGCC-3’(SEQIDNO.33)OLC-VL-F:5’-CTGACCGTGAGCAGCGGTGGCGGTGGCTCGGGCGGTGGTGGGTCGGGTGGCGGCGGATCTGATATTGTGCTGACCCAGAGCC-3’(SEQIDNO.34)VL-R:5’-GCGTTTAATTTCCAGTTTGGTG-3’(SEQIDNO.35)CD8Hinge-F:5’-ACTGGAAATTAAACGCACCACGACGCCAGCGCC-3’(SEQIDNO.36)CD8Hinge-R:5’-GTAGATATCACAGGCGAAGTCCA-3’(SEQIDNO.37)CD8Transmembrane-F:5’-CGCCTGTGATATCTACATCTGGGCGCCCTTGGC-3’(SEQIDNO.38)CD8Transmembrane-R:5’-TCTTTCTGCCCCGTTTGCAGTAAAGGGTGATAACCAGTG-3’(SEQIDNO.39)CD137-F:5’-AAACGGGGCAGAAAGAAACTC-3’(SEQIDNO.40)CD137-R:5’-TGCTGAACTTCACTCTCAGTTCACATCCTCCTTCTTCTTC-3’(SEQIDNO.41)TCR-F:5’-AGAGTGAAGTTCAGCAGGAGCG-3’(SEQIDNO.42)TCR-R:5’-GGAGAGGGGCGTCGACTTAGCGAGGGGGCAGGGC-3’(SEQIDNO.43)WPRE-QPCR-F:5’-CCTTTCCGGGACTTTCGCTTT-3’(SEQIDNO.44)WPRE-QPCR-R:5’-GCAGAATCCAGGTGGCAACA-3’(SEQIDNO.45)Actin-QPCR-F:5’-CATGTACGTTGCTATCCAGGC-3’(SEQIDNO.46)Actin-QPCR-R:5’-CTCCTTAATGTCACGCACGAT-3’(SEQIDNO.47)CAR-QPCR-F:5’-GACTTGTGGGGTCCTTCTCCT-3’(SEQIDNO.48)CAR-QPCR-R:5’-GCAGCTACAGCCATCTTCCTC-3’(SEQIDNO.49)3、SEQIDNO.14、SEQIDNO.15、SEQIDNO.16、SEQIDNO.17、SEQIDNO.18、SEQIDNO.19、SEQIDNO.20、SEQIDNO.21、SEQIDNO.22、SEQIDNO.23、SEQIDNO.24、SEQIDNO.25、SEQIDNO.26、SEQIDNO.27、SEQIDNO.28、SEQIDNO.29、SEQIDNO.30、SEQIDNO.31、、SEQIDNO.32、SEQIDNO.33、SEQIDNO.34、SEQIDNO.35、SEQIDNO.36、SEQIDNO.37、SEQIDNO.38、SEQIDNO.39、SEQIDNO.40、SEQIDNO.41、SEQIDNO.42、SEQIDNO.43、SEQIDNO.44、SEQIDNO.45、SEQIDNO.46、SEQIDNO.47、SEQIDNO.48、SEQIDNO.49所示的DNA序列由上海捷瑞生物工程有限公司合成,并以寡核苷酸干粉或者质粒形式保存;SEQIDNO.52、SEQIDNO.53由常州基宇生物科技有限公司合成;4、工具酶KpnI、PvuI、ApaLI、SacI、ClaI、SalI、T4DNA连接酶均购自NEB公司;5、高保真酶PrimeSTAR、RN购自Takara公司;6、0.22μm-0.8μmPES滤器购自millipore公司;7、质粒抽提试剂盒、琼脂糖凝胶回收试剂盒均购自MN公司;8、感受态细胞TOP10购自tiangen公司;9、NaCl、KCl、Na2HPO4.12H2O、KH2PO4、Trypsin、EDTA、CaCl2、NaOH、PEG6000均购自上海生工;10、Opti-MEM、FBS、DMEM、、1640、Hepes、购自invitrogen公司;11、BiotinylatedproteinL购自GeneScript公司;12、辣根过氧化物酶标记的二抗、DAB工作液均购自北京中杉金桥;13、ECL+plusTMWesternblottingsystem购自Amersham公司;14、DNeasy试剂盒购自上海捷瑞公司;15、淋巴细胞分离液购自深圳达科为公司;16、phycoerythrin(PE)-conjugatedstreptavidin购自BDBioscience公司;17、SA-HRP购自上海翊圣公司;18、支原体检测试剂盒、内毒素检测试剂盒、BCMA+K562细胞购自世翱(上海)公司;19、LDH检测试剂盒购自promega公司;二、重组慢病毒载体lvCAR-BCMA-CLA、lvCAR-BCMA-CLB、lvCAR-BCMA-OLC的构建方法。参见图3,本发明所述重组慢病毒载体的构建方法如下:1、将人EF1α启动子、CD8leader嵌合受体信号肽、BCMA单链抗体重链VH、CommonLinkerA、CommonLinkerB、OptimalLinkerC、BCMA单链抗体轻链VL、CD8Hinge嵌合受体铰链、CD8Transmembrane嵌合受体跨膜区、CD137嵌合受体共刺激因子、TCR嵌合受体T细胞激活域片段克隆至慢病毒骨架质粒pLenti-3Gbasic,分别得到重组慢病毒质粒pCAR-BCMA-CLA,pCAR-BCMA-CLB,pCAR-BCMA-OLC。(1)将慢病毒骨架质粒pLenti-3Gbasic使用ClaI和SalI限制性内切酶进行双酶切,产物经过1.5%的琼脂糖凝胶电泳,确认8303bp的片段V1(图4所示),并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;1、溶胶按200μlNTI/100mggel比例加入溶胶液,50℃水浴放置5-10分钟。2、结合DNA11000g离心30秒,弃去滤液。3、洗膜加入700μlNT3,11000g离心30秒,弃去滤液。4、洗膜重复第三步一次5、晾干11000g离心1分钟,换新的收集管,室温放置1分钟。6、洗脱DNA加入15-30μlNE,室温放置1分钟,11000g离心1分钟,收集滤液。表1琼脂糖凝胶回收步骤(2)用引物EF1α-F和EF1α-R以合成的SEQIDNO.14为模板,使用表2中的体系,PCR循环条件为:98℃3min,(98℃10sec,55℃15sec,72℃2min)*35cycle,72℃10min。产物经过1.5%的琼脂糖凝胶电泳,确认1208bp的片段a,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;试剂体积(μl)H2O32.55×Buffer(withMg2+)10dNTP(各2.5mM)4Primer1(+)(10μM)1Primer2(-)(10μM)1Template1PrimeSTAR0.5表250μlPCR反应体系(3)用引物CD8leader-F和CD8leader-R以合成的SEQIDNO.15为模板,使用表2中的体系,PCR循环条件为:98℃3min,(98℃10sec,55℃15sec,72℃30sec)*35cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认101bp的片段b,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;(4)用引物VH-F和VH-R以合成的SEQIDNO.16为模板,使用表2中的体系,PCR循环条件为:98℃3min,(98℃10sec,55℃15sec,72℃30sec)*35cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认336bp的片段c,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;(5)用引物CLA-VL-F和VL-R以合成的SEQIDNO.20为模板,使用表2中的体系,PCR循环条件为:98℃3min,(98℃10sec,55℃15sec,72℃30sec)*35cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认424bp的片段d,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;(6)用引物CLB-VL-F和VL-R以合成的SEQIDNO.20为模板,使用表2中的体系,PCR循环条件为:98℃3min,(98℃10sec,55℃15sec,72℃30sec)*35cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认430bp的片段e,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;(7)用引物OLC-VL-F和VL-R以合成的SEQIDNO.20为模板,使用表2中的体系,PCR循环条件为:98℃3min,(98℃10sec,55℃15sec,72℃30sec)*35cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认421bp的片段f,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;(8)用引物CD8Hinge-F和CD8Hinge-R以合成的SEQIDNO.21为模板,使用表2中的体系,PCR循环条件为:98℃3min,(98℃10sec,55℃15sec,72℃30sec)*35cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认147bp的片段g,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;(9)用引物CD8Transmembrane-F和CD8Transmembrane-R以合成的SEQIDNO.22为模板,使用表2中的体系,PCR循环条件为:98℃3min,(98℃10sec,55℃15sec,72℃30sec)*35cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认100bp的片段h,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;(10)用引物CD137-F和CD137-R以合成的SEQIDNO.23为模板,使用表2中的体系,PCR循环条件为:98℃3min,(98℃10sec,55℃15sec,72℃30sec)*35cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认142bp的片段i,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;(11)用引物TCR-F和TCR-R以合成的SEQIDNO.24为模板,使用表2中的体系,PCR循环条件为:98℃3min,(98℃10sec,55℃15sec,72℃30sec)*35cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认355bp的片段j,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;(12)将DNA片段b、c、d各1μl作为模板,使用表3中的体系,除引物外加入Eppendorf管内,PCR循环条件为:98℃3min,(98℃10sec,60℃10sec,72℃30sec)*6cycle,加入引物CD8leader-F/VL-R,(98℃10sec,60℃10sec,72℃40sec)*24cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认814bp的片段k,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;试剂体积(μl)H2O33.5-1*模板数5×Buffer(withMg2+)10dNTP(各2.5mM)4Primer1(+)(10μM)1Primer2(-)(10μM)1Template1*模板数PrimeSTAR0.5表350μl重叠PCR反应体系(13)将DNA片段b、c、e各1μl作为模板,使用表3中的体系,除引物外加入Eppendorf管内,PCR循环条件为:98℃3min,(98℃10sec,60℃10sec,72℃30sec)*6cycle,加入引物CD8leader-F/VL-R,(98℃10sec,60℃10sec,72℃40sec)*24cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认820bp的片段l,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;(14)将DNA片段b、c、f各1μl作为模板,使用表3中的体系,除引物外加入Eppendorf管内,PCR循环条件为:98℃3min,(98℃10sec,60℃10sec,72℃30sec)*6cycle,加入引物CD8leader-F/VL-R,(98℃10sec,60℃10sec,72℃40sec)*24cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认811bp的片段m,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;(15)将DNA片段g、h、i、j各1μl作为模板,使用表3中的体系,除引物外加入Eppendorf管内,PCR循环条件为:98℃3min,(98℃10sec,60℃10sec,72℃30sec)*6cycle,加入引物CD8Hinge-F/TCR-R,(98℃10sec,60℃10sec,72℃30sec)*24cycle,72℃5min。产物经过1.5%的琼脂糖凝胶电泳,确认704bp的片段n,并割胶回收置于Eppendorf管内,用MN公司的琼脂糖凝胶回收试剂盒回收相应的片段(见表1),并测定产物的纯度和浓度;(16)将DNA片段V1、a、k、n以5μl总体积且摩尔比1:1:1:1的比例加入Eppendorf管内,加入同源重组酶反应液15μl,混匀后在42℃孵育30分钟,转移至冰上放置2-3分钟,将反应液加入50μlTOP10中,轻轻旋转以混匀内容物,在冰中放置30分钟,将管放到预加温到42℃的恒温水浴锅中热激90秒,快速将管转移到冰浴中,使细胞冷却2-3分钟,每管加900μlLB培养液,然后将管转移到37℃摇床上,温育1小时使细菌复苏,取100μl的转化菌液涂布于AmpLB琼脂平板上,倒置平皿,于恒温培养箱中37℃培养,16小时。挑取克隆进行菌落PCR鉴定,鉴定正确的克隆即为重组慢病毒质粒pCAR-BCMA-CLA,对正确的克隆进行酶切鉴定(见图4);(17)将DNA片段V1、a、l、n以5μl总体积且摩尔比1:1:1:1的比例加入Eppendorf管内,加入同源重组酶反应液15μl,混匀后在42℃孵育30分钟,转移至冰上放置2-3分钟,将反应液加入50μlTOP10中,轻轻旋转以混匀内容物,在冰中放置30分钟,将管放到预加温到42℃的恒温水浴锅中热激90秒,快速将管转移到冰浴中,使细胞冷却2-3分钟,每管加900μlLB培养液,然后将管转移到37℃摇床上,温育1小时使细菌复苏,取100μl的转化菌液涂布于AmpLB琼脂平板上,倒置平皿,于恒温培养箱中37℃培养,16小时。挑取克隆进行菌落PCR鉴定,鉴定正确的克隆即为重组慢病毒质粒pCAR-BCMA-CLB,对正确的克隆进行酶切鉴定(见图5);(18)将DNA片段V1、a、m、n以5μl总体积且摩尔比1:1:1:1的比例加入Eppendorf管内,加入同源重组酶反应液15μl,混匀后在42℃孵育30分钟,转移至冰上放置2-3分钟,将反应液加入50μlTOP10中,轻轻旋转以混匀内容物,在冰中放置30分钟,将管放到预加温到42℃的恒温水浴锅中热激90秒,快速将管转移到冰浴中,使细胞冷却2-3分钟,每管加900μlLB培养液,然后将管转移到37℃摇床上,温育1小时使细菌复苏,取100μl的转化菌液涂布于AmpLB琼脂平板上,倒置平皿,于恒温培养箱中37℃培养,16小时。挑取克隆进行菌落PCR鉴定,鉴定正确的克隆即为重组慢病毒质粒pCAR-BCMA-OLC,对正确的克隆进行酶切鉴定(见图6)。2、重组慢病毒载体lvCAR-BCMA-CLA、lvCAR-BCMA-CLB、lvCAR-BCMA-OLC的包装。(1)完全培养基:取出预热好的新鲜培养基,加入10%FBS+5mlPen-Srep,上下颠倒混匀即可;(2)1XPBS溶液:称量NaCl8g,KCl0.2,Na2HPO4.12H2O3.58g,KH2PO40.24g置于1000ml烧杯中,加入900mlMilli-Qgrade超纯水溶解,溶解完成后,使用1000ml量筒定容至1000ml,121℃高温湿热灭菌20min;(3)0.25%Trypsin溶液:称量Trypsin2.5g,EDTA0.19729g置于1000ml烧杯中,加入900ml1XPBS溶解,溶解完成后,使用1000ml量筒定容至1000ml,0.22μM过滤除菌,长期使用可保存至-20℃冰箱;(4)0.5MCaCl2溶液:称量36.75gCaCl2用400mlMilli-Qgrade超纯水溶解;用Milli-Qgrade超纯水将总体积定容至500ml,混匀;0.22μm过滤除菌,分装保存到50ml离心管中,每管45ml左右,4℃保存。(5)2XHBS溶液:称量4.09gNaCl,0.269gNa2HPO4,5.96gHepes,用400mlMilli-Qgrade超纯水溶解;校准pH仪后,用2MNaOH溶液将HBS溶液的pH调到7.05。调整每瓶HBS的PH消耗2MNaOH为3ml左右;(6)从液氮罐中取出冻存的HEK293T/17细胞,迅速转移到37℃水浴中,1~2min后转移到超净台中,无菌操作将冻存管中的液体全部转移至10cm2培养皿中,补足含10%FBS的DMEM至8mL/10cm2dish,24h后显微镜观察细胞,细胞汇合的程度大于80%进行传代;(7)选择细胞状态良好、无污染的HEK293T/17细胞,每2-6个培养皿为一组,将细胞胰酶消化后,用电动移液器吸取4-12ml完全培养基,向每个消化后的培养皿中加2ml,避免培养皿变干;使用1ml移液器将所有细胞吹打成单细胞悬液,转移到培养基瓶中;(8)将上述2-6个培养皿中的剩余细胞转移到培养基瓶中,并用培养基再冲洗一便培养皿;(9)盖紧培养基瓶盖,上下颠倒10次左右充分混匀细胞悬液,将细胞传到8-24个10cm2培养皿中,每皿的细胞密度应当约4×106个/10ml完全培养基左右。如果细胞密度和预期的相差较大,则需要对细胞进行计数,然后按照4×106个/皿的量接种;(10)每6个培养皿整理为一摞,注意保持上下皿之间的配合。将培养皿左右,前后晃动数次,使细胞充分铺开,然后放入5%CO2培养箱。剩余细胞做同样处理;(11)检查所传代细胞,细胞汇合度应当为70-80%,轮廓饱满,贴壁良好,在细胞培养皿中均匀分布;(12)为细胞换液,将培养基替换为新鲜完全培养基,每皿9ml,并将培养箱的CO2浓度设定值提高到8%;(13)按照N+0.5配DNA/CaCl2溶液。每皿HEK293T/17细胞转染质粒量按照下列比例使用:重组慢病毒质粒(20μg),pPac-GP(15μg),pPac-R(10μg),pEnv-G(7.5μg)。取一个新的5ml离心管,加入0.5MCaCl2:0.25ml,重组慢病毒质粒20μg:pPac-GP15μg:pPac-R10μg:pEnv-G7.5μg,补充超纯水至0.5ml盖上盖子,充分混匀;(14)另取一支5ml离心管,加入0.5mlDNA/CaCl2溶液。打开涡旋振荡器,一只手拿住5ml离心管的上端,使管底接触振荡头,使液体在管壁上散开流动,另一只手拿一把1mL移液枪,吸取0.5mL2×HBS溶液,缓慢滴加进入离心管,控制流速,以半分钟滴完为宜。2×HBS加入后,继续振荡5秒钟,停止振荡,可直接加入需要转染的细胞中;(15)取一皿细胞,将离心管中的1mL钙转液滴加进去,尽可能使钙转试剂分布到整个培养皿中;(16)钙转液加入后,在皿盖上做好标记,将培养皿放还到另一个5%CO2培养箱中。确保培养皿水平放置,每摞培养皿不要超过6个。在5%CO2培养箱中放置(6–8h);(17)将第一个培养箱的CO2浓度设定值调回到5%;(18)24小时后,检查细胞状态。细胞汇合度应当为80–85%左右,状态良好。将培养基吸走,更换10ml新鲜的DMEM完全培养基;(19)48小时后,观察转染效率。绝大多数细胞仍然是贴壁的。可以看到超过95%细胞都会带有绿色荧光。将同一个病毒包装上清液收集到一起,并向培养皿中继续添加10mL新鲜培养基;(20)72小时后,再次将同一个病毒上清液收集到一起,两次收集的病毒可以放在一起,丢弃培养皿;此时收集的上清里包含了重组慢病毒载体lvCAR-BCMA-CLA、lvCAR-BCMA-CLB、lvCAR-BCMA-OLC。实施例2重组慢病毒载体的浓缩及检测一、超速离心法纯化重组慢病毒载体;(1)将收集的上清液分装到50ml离心管中,500g室温离心10min,除去细胞和大的碎片;(2)用0.22μm-0.8μm滤器过滤上清液;(3)取6个Hitachi40PA超速离心管,表面喷洒70%乙醇消毒,放在超净台用紫外灯照射杀菌30分钟。也可以通过高温湿热灭菌;(4)将第2步处理好的细胞上清样品分装32ml到离心管中;(5)盖上金属盖,将离心管连同金属盖一起配平,用1XPBS调整使重量偏差在0.02g范围内;(6)将配平的离心管对称放置于超速离心转子P50AT2中。设置离心转速100,000g,4℃离心2小时;(7)离心结束后,小心将离心管从转子中拿出,可以看到在离心管底有一小团沉淀,用Marker笔在外管壁上做标记,倒掉上清。将离心管倒扣在预先铺好的纸巾上,使残余液体流掉。可以用移液枪将挂在壁上的液滴吸走;(8)向每个离心管中加入200μl的Opti-MEM,用200μl移液器吹打使沉淀溶解,尽量减少泡沫的产生;(9)将超速离心管插入50ml离心管中,盖上盖子,放入4℃冰箱过夜;(10)500g,室温离心1min,使病毒液集中到管底;(11)将所有相同的病毒浓缩液集中到一起,用0.22μm-0.8μm的PES滤器过滤;将病毒分成25到50μl一管,冻存到-80℃冰箱,进行长期保存;二、高速离心法纯化重组慢病毒载体;(1)将收集的上清液204ml使用0.22μm-0.8μm的PES滤器过滤;(2)加入51ml50%的PEG6000溶液;(3)加入21.7ml4MNaCl溶液;(4)加入23.3ml的PBS溶液,这时溶液总体积为300ml.PEG6000的终浓度8.5%、NaCl终浓度0.3M;(5)将溶液分装进250ml广口瓶中,每份150ml;(6)4℃放置1.5小时,每20-30分钟混匀一次;(7)4℃、7000g离心10min;(8)离心后能看见管底有白色沉淀;(9)小心弃去上清液,每瓶加入1.2ml50mMTris-HCl(pH7.4),剧烈摇晃重悬沉淀;(10)涡旋震荡20–30秒进一步重悬沉淀;(11)将病毒分成25到50μl一管,冻存到-80℃冰箱,进行长期保存;三、离子交换色谱法纯化重组慢病毒载体(如图7所示);(1)将收集的上清液使用Thermo真空泵,经0.22μm-0.8μm的PES滤器抽滤,除去杂质;(2)按1:1~1:10的比例往上清中加入1.5MNaCl250mMTris-HCl(pH6-8);(3)将2个离子交换柱串联放置,用4ml1MNaOH、4ml1MNaCl、5ml0.15MNaCl25mMTris-HCl(pH6-8)溶液依次过柱;(4)将步骤2中获得的溶液通过蠕动泵以1-10ml/min的速度给离子交换柱上样;(5)全部上清液过柱后,使用10ml0.15MNaCl25mMTris-HCl(pH6-8)溶液清洗一遍;(6)根据上样量使用1-5ml1.5MNaCl25mMTris-HCl(pH6-8)进行洗脱,收集洗脱液;(7)将洗脱液分成25到50μl一管,冻存到-80℃冰箱,进行长期保存;四、滴度测定及比较;(1)取24孔板接种293T细胞。每孔细胞为5×104个,所加培养基体积为500ul,不同种类的细胞生长速度有所差异,进行病毒感染时的细胞融合率为40%-60%;(2)准备3个无菌EP管,在每个管中加入90ul的新鲜完全培养基(高糖DMEM+10%FBS)接种细胞24小时后,取两个孔的细胞用血球计数板计数,确定感染时细胞的实际数目,记为N;(3)取待测定的病毒原液10ul加入到第一个管中,轻轻混匀后,取10ul加入到第二个管中,然后依次操作直到最后一管;在每管中加入410ul完全培养基(高糖DMEM+10%FBS),终体积为500ul;(4)感染开始后20小时,除去培养上清,更换为500μl完全培养基(高糖DMEM+10%FBS),5%CO2继续培养48小时;(5)72小时后,观察荧光表达情况,正常情况下,荧光细胞数随稀释倍数增加而相应减少,并拍照;(6)用0.2ml0.25%胰酶-EDTA溶液消化细胞,在37℃放置1分钟。用培养基吹洗整个细胞面,离心收集细胞。按照DNeasy试剂盒的说明抽提基因组DNA。每个样品管中加入200μl洗脱液洗下DNA并定量;(7)准备目的DNA检测qPCRmix总管Ⅰ(QPCR引物序列为SEQIDNO.44---SEQIDNO.45):n=numberofreactions.例如:总反应数为40,将1ml2×TaqManUniversalPCRMasterMix,4μlforwardprimer,4μlreverseprimer,4μlprobe和788μlH2O混和。震荡后放在冰上;(8)准备内参DNA检测qPCRmix管Ⅱ(QPCR引物序列为SEQIDNO.46---SEQIDNO.47):2×TaqManMasterMix25μl×n10×RNasePprimer/probemix2.5μl×nH2O17.5μl×nn=numberofreactions.例如:总反应数为40,将1ml2×TaqManUniversalPCRMasterMix,100μl10×RNasePprimer/probemix和700μlH2O混和。震荡后放在冰上;(9)在预冷的96孔PCR板上完成PCR体系建立。从总管Ⅰ中各取45μl加入到A-D各行的孔中,从总管Ⅱ中各取45μl加入到E-G各行的孔中。(10)分别取5μl质粒标准品和待测样品基因组DNA加入到A-D行中,每个样品重复1次。另留1个孔加入5μl的水做为无模板对照(no-templatecontrol)。(11)分别取5μl基因组标准品和待测样品基因组DNA加入到E-G行中,每个样品重复1次。另留1个孔加入5μl的水做为无模板对照(no-templatecontrol)。(12)所使用定量PCR仪为ABIPRISM7500定量系统。循环条件设定为:50℃2分钟,95℃10分钟,然后是95℃15秒,60℃1分钟的40个循环。数据分析:测得的DNA样品中整合的慢病毒载体拷贝数用基因组数加以标定,得到每基因组整合的病毒拷贝数。滴度(integrationunitsperml,IUml-1)的计算公式如下:IUml-1=(C×N×D×1000)/V其中:C=平均每基因组整合的病毒拷贝数N=感染时细胞的数目(约为1×105)D=病毒载体的稀释倍数V=加入的稀释病毒的体积数(13)重组慢病毒载体lvCAR-BCMA-CLA、lvCAR-BCMA-CLB、lvCAR-BCMA-OLC的滴度结果(如图8所示),离子交换色谱法的结果明显优于超速离心法和高速离心法;五、内毒素测定及比较;(1)、内毒素工作标准品为15EU/支;(2)、鲎试剂灵敏度λ=0.25EU/ml,0.5ml/管(3)、内毒素标准品稀释:取内毒素标准品一支,分别用BET水按比例稀释成4λ和2λ的溶解,封口膜封口,震荡溶解15min;稀释时每稀释一步均应在漩涡混合器上混匀30s;(4)、加样:取鲎试剂若干支,每支加入BET水0.5ml溶解,分装至若干支无内毒素试管中,每管0.1ml。其中2支为阴性对照管,加入BET水0.1ml;2支为阳性对照管,加入2λ浓度的内毒素工作标准品溶液0.1ml;2支为样品阳性对照管,加入0.1ml含2λ内毒素标准品的样品溶液(稀释20倍的待测样品1ml+4λ的内毒素标准品溶液1ml=2ml含2λ内毒素标准品的稀释40倍样品)。样品管中加入0.1ml样品,稀释比例见表4,37±1℃水浴(或培养箱)保温60±1min;(5)、重组慢病毒载体lvCAR-BCMA-CLA、lvCAR-BCMA-CLB、lvCAR-BCMA-OLC的内毒素检测结果(如表4所示),超速离心法的内毒素含量在20~40EU/ml之间,高速离心法的内毒素含量在20~40EU/ml之间,离子交换色谱法的内毒素含量在1.25~2.5EU/ml之间明显优于超速离心法和高速离心法;表4重组慢病毒载体的不同纯化方式的内毒素检测结果六、支原体测定及比较;(1)在实验前三日,细胞样品用无抗生素培养基进行培养;(2)收集1ml细胞悬浮液(细胞数大于1*105),置于1.5ml离心管中;(3)13000×g离心1min,收集沉淀,弃去培养基;(4)加入500ulPBS用枪头吹吸或涡旋振荡,重悬沉淀。13000×g离心5min;(5)步骤4重复一次;(6)加入50μlCellLysisBuffer,用枪头吹吸,充分混匀后,在55℃水浴中孵育20min;(7)将样品置于95℃中加热5min;(8)13000×g离心5min后,取5μl上清作为模板,25μlPCR反应体系为:ddH206.5μl、MycoMix1μl、2xTaqPlusMixMaster(DyePlus)12.5μl、模板5μl;PCR循环条件为:95℃30sec,(95℃30sec,56℃30sec,72℃30sec)*30cycle,72℃5min。(9)支原体检测结果显示(如图9和表5所示),超速离心法、高速离心法、离子交换色谱法纯化的重组慢病毒载体均不含支原体。表5支原体检测实施例3重组慢病毒载体lvCAR-BCMA-CLA、lvCAR-BCMA-CLB、lvCAR-BCMA-OLC的功能检测一、CAR基因的细胞水平表达检测:(1)重组慢病毒载体lvCAR-BCMA-CLA、lvCAR-BCMA-CLB、lvCAR-BCMA-OLC感染PBMC细胞后,收集细胞采用RT-PCR进行CARmRNA转录水平的检测,验证CAR基因的表达,如果CARmRNA转录水平增高,则说明CAR基因的转录水平表达成功;(2)重组慢病毒载体lvCAR-BCMA-CLA、lvCAR-BCMA-CLB、lvCAR-BCMA-OLC感染PBMC细胞后,收集细胞采用westernblot进行CAR蛋白表达水平的检测,验证CAR基因的表达,如果CAR蛋白表达水平增高,则说明CAR基因的翻译水平表达成功;(3)分别将MOI=15的lvCAR-BCMA-CLA、lvCAR-BCMA-CLB、lvCAR-BCMA-OLC和对照病毒MOCK感染细胞,48h后提取6孔板中细胞的总RNA和总蛋白分别进行荧光定量PCR实验和免疫印迹实验。具体步骤:包被6孔板的四个孔,每个孔加入相应的PBS和RN,4℃过夜。12小时后按MOI=15包被病毒,37℃培养箱放置5h;取出的6孔板,弃掉病毒上清,用PBS洗两遍,按1*106/孔,包被PBMC(用淋巴细胞分离液从人血中分离),加入500ul培养基(含10%血清、20U/mlIL-2、Polybrene8ug/ml)。静置20min,1000g20℃离心30min,37℃培养48h。(4)Trizol法提取6孔板中PBMC细胞的总RNA,逆转录扩增cDNA,用QPCR引物(序列为SEQIDNO.46---SEQIDNO.49)进行荧光定量PCR实验,反应体系见表6,以内参Actin为对照组,验证其mRNA的转录情况。试剂体积(μl)SYBRpremixextaq:10μlROXReverseDye(50x)0.4μl上游引物(2.5μM):0.5μl下游引物(2.5μM):0.5μlcDNA1.0μlddH2O7.6μl表620μlqPCR反应体系(5)蛋白免疫印迹(WesternBlot)通过聚丙烯酰胺凝胶电泳将从PBMC中提取的总蛋白质按相对分子质量分离。采用湿转(4℃,400mA,120min),将蛋白转移到PVDF膜上。用封闭液(含5%脱脂牛奶的TBST溶液)室温封闭PVDF膜1h,封闭液1:1000稀释BiotinylatedproteinL,然后与封闭好的PVDF膜室温孵育4℃过夜。TBST洗膜3次,每次10min。封闭液1:500稀释相应的SA-HRP,室温下孵育PVDF膜2h,TBST洗膜3次,每次10min。采用Amersham公司ECL+plusTMWesternblottingsystem试剂盒进行显色。X光显影获得显示条带的胶片。(6)RT-QPCR检测显示,重组慢病毒载体感染PBMC后的CAR的表达水平比对照病毒MOCK和空细胞有明显升高(如图10和表7所示),说明CAR基因的转录水平表达成功。表7RT-QPCR检测(7)蛋白免疫印迹(WesternBlot)的结果表明,CAR蛋白在重组慢病毒系统中表达(如图11所示),说明CAR基因的翻译水平表达成功。二、细胞因子分泌及杀伤效果评估。(1)分别培养BCMA+K562细胞和效应细胞lvCAR-BCMA-CLA-PBMC、lvCAR-BCMA-CLB-PBMC、lvCAR-BCMA-OLC-PBMC;(2)收集靶细胞(BCMA+K562)4x105cells和效应细胞(CART细胞)2.8x106cells,800g,6min离心,弃上清;(3)用1ml1xPBS溶液分别重悬靶细胞和效应细胞,800g,6min离心,弃上清;(4)重复步骤3一次;(5)用700ul培养基(1640培养基+10%FBS)重悬效应细胞,用2ml培养基(1640培养基+10%FBS)重悬靶细胞;(6)设置效靶比为1:1、5:1、10:1的实验孔,并设置对照组,每组3个复孔;(7)250xg,5min平板离心;(8)37℃5%CO2培养箱中培养4小时;(9)250xg,5min平板离心;(10)取每个孔的50ul上清到新96孔板中,并且每孔加入50ul底物溶液(避光操作);(11)避光孵育25分钟;(12)每孔加入50ul终止液;(13)酶标仪检测490nm吸光度;(14)将3个复孔取平均值;将所有实验孔、靶细胞孔和效应细胞孔的吸光值减去培养基背景吸光值的均值;将靶细胞最大值的吸光值减去体积校正对照吸光值的均值。(15)将步骤14中获得的经过校正的值带入下面公式,计算每个效靶比所产生的细胞毒性百分比。结果如图12所示,重组慢病毒载体lvCAR-BCMA-OLC转导的PBMC细胞在不同效靶比条件下杀伤效率明显高于lvCAR-BCMA-CLA和lvCAR-BCMA-CLB转导的PBMC细胞;杀伤效率=(实验孔-效应细胞孔-靶细胞孔)/(靶细胞最大孔-靶细胞孔)X100%(16)重复准备一份步骤6的样品用于qPCR检测细胞因子表达水平,结果如图13所示,重组慢病毒载体lvCAR-BCMA-OLC转导的PBMC细胞在不同效靶比条件下IL-2和IFN-γ的mRNA转录水平明显高于lvCAR-BCMA-CLA和lvCAR-BCMA-CLB转导的PBMC细胞;三、根据文献报道(B-cellMaturationAntigenisaPromisingTargetforAdoptiveTcellTherapyofMultipleMyeloma.RobertO.Carpenter,etal.ClinCancerRes.2013April15;19(8):2048–2060.),CAR-BCMA载体针对多发性骨髓瘤细胞有着良好的效果。本研究使用表达CAR-BCMA基因的T淋巴细胞,研究其在体外对多发性骨髓瘤细胞系的杀伤作用以及回输小鼠体内对肿瘤体积以及生存期的影响。CAR-BCMA-T淋巴细胞对H929多发性骨髓瘤细胞系与对照相比有明显的杀伤作用(如图14A所示);CAR-BCMA-T淋巴细胞对RPMI8226多发性骨髓瘤细胞系与对照相比有明显的杀伤作用(如图14B所示);CAR-BCMA-T淋巴细胞回输小鼠后,15天内对肿瘤的抑制效果明显(如图14C所示);CAR-BCMA-T淋巴细胞回输小鼠后,小鼠的生存期明显延长(如图14D所示)。结果显示,过继性CAR-BCMA-T细胞疗法在针对多发性骨髓瘤的治疗方面拥有良好的前景。实施例4第三代慢病毒骨架载体3rdLV-CAR-BCMA与第二代慢病毒骨架载体2ndLV-CAR-BCMA滴度比较分别构建3rdLV-CAR-BCMA和2ndLV-CAR-BCMA;质粒构建、病毒包装、病毒超速离心浓缩、滴度测定过程参考实施例1;连续3个批次的滴度结果如图15所示,第三代慢病毒骨架载体3rdLV-CAR-BCMA的滴度要优于第二代慢病毒骨架载体2ndLV-CAR-BCMA;因此,本专利选择使用第三代慢病毒骨架载体作为CAR基因的搭载骨架。实施例5第二代CAR慢病毒载体3rdLV-2ndCAR-BCMA与第三代CAR慢病毒载体3rdLV-3rdCAR-BCMA杀伤效率比较分别构建3rdLV-2ndCAR-BCMA和3rdLV-3rdCAR-BCMA;质粒构建、病毒包装、病毒超速离心浓缩、滴度测定过程参考实施例1;杀伤实验参考实施例3;第二代CAR慢病毒载体3rdLV-2ndCAR-BCMA与第三代CAR慢病毒载体3rdLV-3rdCAR-BCMA杀伤效率结果比较如图16所示,3rdLV-3rdCAR-BCMA针对靶细胞的杀伤效率在不同效靶比条件下并未表现的比3rdLV-2ndCAR-BCMA更高效;因此,本专利选择使用第二代CAR设计的慢病毒载体作为临床实验载体。以上已对本发明创造的较佳实施例进行了具体说明,但本发明创造并不限于所述实施例,熟悉本领域的技术人员在不违背本发明创造精神的前提下还可作出种种的等同的变型或替换,这些等同的变型或替换均包含在本申请权利要求所限定的范围内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1