一种化合物A苯甲酸盐的无定形及其制备方法和含有该无定形的药物组合物与流程
本发明属于药物化合物的制备领域,具体涉及一种化合物A苯甲酸盐的无定形及其制备方法,以及含有该无定形的药物组合物。
背景技术:
二肽基肽酶IV(Dipeptidyl peptidase IV,DPP-IV)是丝氨酸蛋白酶,能特异性水解多肽或蛋白质N末端的Xaa-Pro或Xaa-Ala二肽。DPP-IV是非典型丝氨酸蛋白酶,其C末端区域的Ser-Asp-His催化三联体与典型丝氨酸蛋白酶不同,为逆序排列。DPP-IV为II型膜整合蛋白,广泛分布于哺乳动物各组织。DPP-IV在分化上皮细胞表面表达,如肠、肝脏、肾近端小管、前列腺、黄体和白细胞亚型如淋巴细胞和巨噬细胞。血清中存在DPP-IV的可溶性蛋白形式,其结构和功能与膜结合蛋白形式相同但缺少疏水跨膜结构域。
DPP-IV有多种生理学相关底物,如炎症趋化因子类、正常T细胞表达和分泌因子(regulated on activation normal T-cell expressed and secreted,RANTES)、嗜酸细胞活化趋化因子和巨噬细胞衍生趋化因子、神经肽类如神经肽Y(neuropeptide Y,NPY)和P5物质、血管活性肽、肠降血糖素如胰高糖素样肽(glucagon-like peptide-l,GLP-1)和葡萄糖依赖性促胰岛素多肽(glucose-dependent insulinotropic polypeptide,GIP)。
抑制体内DPP-IV可使内源性GLP-1(7-36)水平上升,减少其拮抗物GLP-1(9-36)的生成。因此,DPP-IV抑制剂可能对与DPP-IV活性相关的疾病有效,例如2型糖尿病,糖尿病血脂异常,糖耐量降低(impaired glucose tolerance,IGT),空腹血糖受损(impaired fasting plasma glucose,IFG),代谢性酸中毒,酮病,食欲调节和肥胖。
因此,抑制DPP-IV被认为是新的治疗II型糖尿病的治疗途径。
WO2011079778描述了DPP-IV可逆的竞争性抑制剂化合物,其化学名称为:(R)-2-((3-(3-氨基哌啶-1-基)-6-甲基-5-氧代-1,2,4-三嗪-4(5H)-基)甲基)-4-氟苄腈(以下简称化合物A),分子式:C17H19FN6O,分子量:342,化学结构式为下式(I):
为了改善该化合物的药用性质,对具有有利的稳定性性质研究可以有效地用于药物组合物中通过抑制DPP-IV而治疗病态的患者。
技术实现要素:
本发明的目的之一在于提供一种稳定的二肽基肽酶-IV可逆的竞争性抑制剂化合物A苯甲酸盐的无定形。
化合物A苯甲酸盐的化学名称为:(R)-2-((3-(3-氨基哌啶-1-基)-6-甲基-5-氧代-1,2,4-三嗪-4(5H)-基)甲基)-4-氟苄腈苯甲酸盐(即化合物A苯甲酸盐),分子式:C17H19FN6O·C7H6O2,分子量:464.49,化学结构式为下式(II),
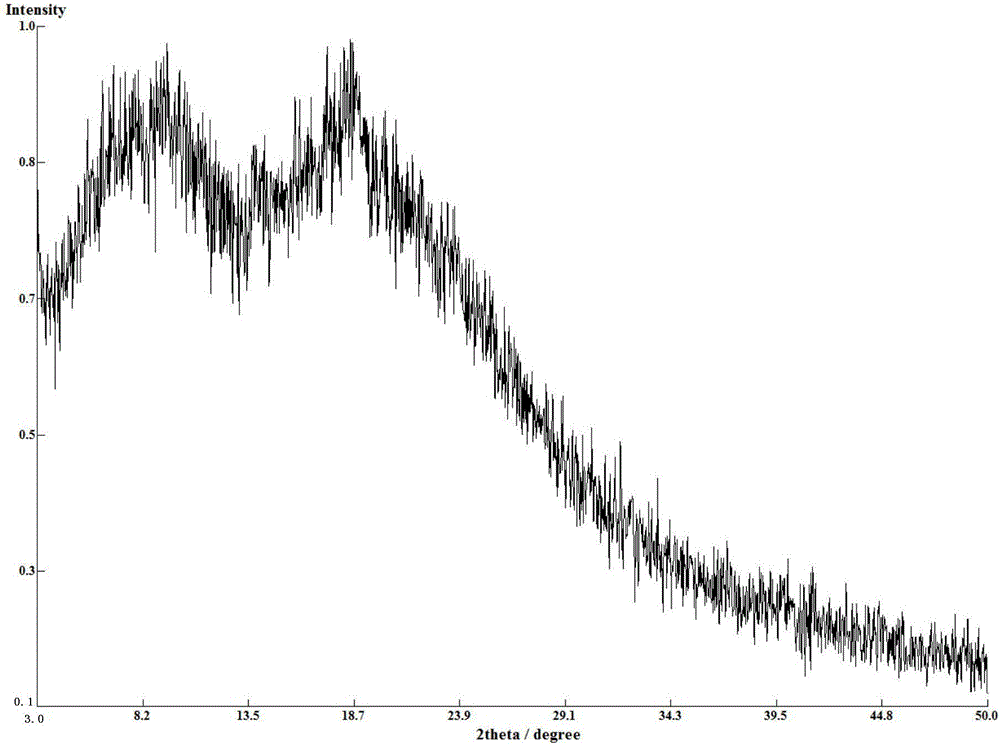
上述化合物A苯甲酸盐的无定形在X射线衍射图中无晶体特征吸收峰。
更为具体的,所述化合物A苯甲酸盐的无定形的X射线衍射图如附图1或附图2所示。
X射线衍射检测条件:采用锐影(Empyrean)X射线衍射仪,在Cu靶Kα射线,电压:40.0kV,电流:40.0mA,步长0.02°条件下测定2θ范围:3°-50°。
上述化合物A苯甲酸盐的无定形,其差热分析图谱中无明显吸热峰。
更为具体的,所述化合物A苯甲酸盐的无定形的差热分析图谱如附图3或附图4所示。
差热分析检测条件:采用德国NETZSCH公司DSC 200F3差示扫描量热仪,气氛:N2(纯度:≥99.99%),20mL/min。
上述化合物A苯甲酸盐的无定形,其具有如附图5所示的TG图谱。
热重分析检测条件:采用德国NETZSCH公司TG 209F3热重分析仪,气氛:N2(纯度:≥99.99%)。
所述化合物A苯甲酸盐的无定形具有高稳定性,在高湿条件下保存,含量基本不发生变化。并且,化合物A苯甲酸盐的无定形相对于化合物A具有更好的稳定性,充分保证用药的安全性和有效性。
本发明的另一目的在于提供一种上述化合物A苯甲酸盐的无定形的制备方法,该方法工艺简单,常温条件下即可实现。
将化合物A苯甲酸盐置于有机溶剂中,搅拌溶解,20℃~30℃减压浓缩,得气泡样物品,常温真空干燥,制备得到化合物A的无定形。
所述的有机溶剂为二氯甲烷、乙腈、乙酸乙酯、甲醇、正己醇中的一种或两种混合。
其中化合物A可根据WO2011079778公开的方法制备,具体合成路线及主要的反应条件如下:
化合物A苯甲酸盐的制备方法如下:将化合物A进行精制,以有机溶剂或有机溶剂与水的混合溶液作为溶剂,分别溶解苯甲酸和和精制后的化合物A,内温保持在10℃~35℃下,化合物A溶液中滴加等摩尔的苯甲酸溶液,所得反应液洗涤过滤,浓缩干燥,即得化合物A苯甲酸盐。所述的有机溶剂为乙醇、四氢呋喃或其两种任意比例的混合物。
本发明的再一目的在于提供一种含有上述的化合物A苯甲酸盐无定形的药物组合物,以及一种以上药学上可接受的载体。
所述载体包括各种药用辅料,包材,传递工具等,根据制剂需要进行选择,例如辅料包括崩解剂、粘合剂、润滑剂等,可以适用于口服、吸入、非肠胃给药或表面使用;剂型包括但不限于注射剂、溶液制剂、片剂、胶囊剂、颗粒剂等。
所述药物组合物可以用于制备DPP-IV引起相关疾病、特别是2型糖尿病的药物的应用。
本发明与现有技术相比具有如下突出的优点及有益效果:
1、本发明的化合物A苯甲酸盐的无定形的纯度高,易于药物组合物的配置和使用。
2、本发明的化合物A苯甲酸盐的无定形的稳定性高,有利于药物组合物的制备和使用。
3、本发明制备化合物A苯甲酸盐的无定形的方法简单、快捷、在常温条件下即可制备,易于产业化生产。
附图说明
图1是本发明化合物A苯甲酸盐的无定形实施例3的X射线衍射图谱
图2是本发明化合物A苯甲酸盐的无定形实施例4的X射线衍射图谱
图3是本发明化合物A苯甲酸盐的无定形实施例3的DSC图谱
图4是本发明化合物A苯甲酸盐的无定形实施例4的DSC图谱
图5是本发明化合物A苯甲酸盐的无定形实施例3的TG图谱
具体实施方式
下面结合实施例和附图对本发明作进一步详细的描述,但发明的实施方式不限于此。
实施例1化合物A的制备
按照WO2011079778说明书实施例2和3的方法,采用以下技术合成路线制备化合物A:
所得化合物A,1H-NMR(400MHz,DMSO,ppm):δ7.96(m,1H),7.36(br,1H),7.29(d,1H),5.23(s,2H),3.15(m,3H),2.72(m,2H),2.23(s,3H),1.78(d,1H),1.64(d,1H),1.47(m,1H),1.12(m,1H).MS:m/z,343(100%,M+1)。
具体制备步骤如下:
步骤A.1-溴-4-氟-2-(异硫氰酸甲基)苯(2)
向1-溴-2-(溴甲基)-4-氟苯(1,5.36g,20.0mmol)的DMF溶液(20mL)中,加入碘化钠(1.20g,8.00mmol)和硫氰酸钾(3.88g,40.0mmol)。该混合物在氮气氛围下加热到80℃反应12h后,冷却到室温,向其中加入100mL水,并用乙酸乙酯萃取(50mL×2),合并有机层用饱和食盐水洗涤,无水硫酸镁干燥,抽滤浓缩得粗品,残余物用硅胶柱色谱纯化(洗脱剂:石油醚)得1-溴-4-氟-2-(异硫氰酸甲基)苯(2)。
步骤B.N-(2-溴-5-氟苯甲基)肼基硫代甲酰胺(3)
将水合肼(80%,2.22g,35.5mmol)的1,4-二氧六环溶液(20mL)冷却到0℃,向其中加入1-溴-4-氟-2-(异硫氰酸甲基)苯(2,3.16g,12.8mmol)的1,4-二氧六环溶液(5mL)。该混合物在室温搅拌2h,向其中加入100mL冰水,有固体析出,抽滤,水洗,五氧化二磷干燥过夜,得N-(2-溴-5-氟苯甲基)肼基硫代甲酰胺(3)。MS:m/z,278(100%,M+1),280(100%),300(10%,M+23),302(10%)。
步骤C.甲基2-(2-(2-溴-5-氟苯甲氨基硫代甲酰胺)肼基)丙酸酯(4)
向丙酮酸(352mg,4.00mmol)的甲醇溶液(15mL)中先后加入N-(2-溴-5-氟苯甲基)肼基硫代甲酰胺(3,1.112g,4.00mmol),以及浓硫酸5滴,将该混合物加热到回流7h,蒸除大部分溶剂,残余物用乙酸乙酯萃取(150mL),有机层先后分别用水,饱和碳酸氢钠溶液,饱和食盐水洗涤,无水硫酸镁干燥,抽滤浓缩得甲基2-(2-(2-溴-5-氟苯甲氨基硫代甲酰胺)肼基)丙酸酯(4)。MS:m/z,362(100%,M+1),364(100%),384(60%,M+23),386(60%)。
步骤D.4-(2-溴-5-氟苯甲基)-6-甲基-3-硫代-3,4-二氢-1,2,4-三嗪-5(2H)-酮(5)
将由钠(273mg,11.88mmol)和干燥甲醇(30mL)新鲜制备的甲醇钠(0.4M)溶于甲醇30mL,向其中加入甲基2-(2-(2-溴-5-氟苯甲氨基硫代甲酰胺)肼基)丙酸酯(4,1.434g,3.96mmol),将该混合物加热回流22h,蒸除大部分溶剂,残余物用水100mL稀释,用2N浓盐酸调节pH为1~2,乙酸乙酯萃取(50mL×2),合并萃取层用饱和食盐水洗涤,无水硫酸钠干燥,抽滤浓缩得粗品,经硅胶柱色谱纯化(洗脱剂:乙酸乙酯/石油醚=20%~30%),得4-(2-溴-5-氟苯甲基)-6-甲基-3-硫代-3,4-二氢-1,2,4-三嗪-5(2H)-酮(5),MS:m/z,330(65%,M+1),332(60%,M+23)。
步骤E.4-(2-溴-5-氟苯甲基)-6-甲基-3-(甲硫基)-1,2,4-三嗪-5(4H)-酮(6)
将4-(2-溴-5-氟苯甲基)-6-甲基-3-硫代-3,4-二氢-1,2,4-三嗪-5(2H)-酮(5,914mg,2.77mmol)悬浮于乙醇15mL中,先后加入氢氧化钠(111mg,2.77mmol)和碘甲烷(787mg,5.54mmol)。将该混合物于室温搅拌10分钟得澄清黄色溶液,反应用水100mL稀释,乙酸乙酯萃取(30mL×2),合并萃取层用饱和食盐水洗涤,无水硫酸镁干燥,抽滤浓缩,残余物用硅胶柱色谱纯化(洗脱剂:乙酸乙酯/石油醚=20~25%)得4-(2-溴-5-氟苯甲基)-6-甲基-3-(甲硫基)-1,2,4-三嗪-5(4H)-酮(6).1H NMR(400MHz,DMSO,ppm):δ7.73(m,1H),7.16(br,1H),7.05(d,1H),5.09(s,2H),2.56(s,3H),2.32(s,3H).MS:m/z,344(100%,M+1),346(100%)。
步骤F.(R)-叔丁基1-(4-(2-溴-5-氟苯甲基)-6-甲基-5-氧代-4,5-二氢-1,2,4-三嗪-3-基)哌啶-3-氨基甲酸酯(8)
将4-(2-溴-5-氟苯甲基)-6-甲基-3-(甲硫基)-1,2,4-三嗪-5(4H)-酮(6,180mg,0.523mmol)与(R)-叔丁基哌啶-3-氨基甲酸酯(7,208mg,1.04mmol)研磨5分钟,在氮气氛围下加热到135℃反应13h,反应混合物用硅胶柱色谱纯化(洗脱剂:乙酸乙酯/石油醚=10~50%)得(R)-叔丁基1-(4-(2-溴-5-氟苯甲基)-6-甲基-5-氧代-4,5-二氢-1,2,4-三嗪-3-基)哌啶-3-氨基甲酸酯(8).MS:m/z,496(100%,M+1),498(100%)。
步骤G.(R)-叔丁基1-(4-(2-氰基-5-氟苄基)-6-甲基-5-氧代-4,5-二氢-1,2,4-三嗪-3-基)哌啶-3-氨基甲酸酯(9)
向碳酸钠(53mg,0.50mmol)、醋酸钯(3mg,0.013mmol)和N-甲基吡咯烷酮0.5mL的混合物中加入异丙醇3滴和水2滴,该混合物室温搅拌5分钟,向其中加入(R)-叔丁基1-(4-(2-溴-5-氟苯甲基)-6-甲基-5-氧代-4,5-二氢-1,2,4-三嗪-3-基)哌啶-3-氨基甲酸酯(8,246mg,0.496mmol)的NMP溶液(1.0mL),并加热到140℃,再加入K4[Fe(CN)6].3H2O(209mg,0.496mmol),在140℃加热12h,冷却到室温,加入水10mL,乙酸乙酯萃取(20mL×2),合并有机层用饱和食盐水洗涤,无水硫酸镁干燥,抽滤浓缩得粗品,经硅胶柱色谱纯化(洗脱剂:乙酸乙酯/石油醚=20~35%)得(R)-叔丁基1-(4-(2-氰基-5-氟苄基)-6-甲基-5-氧代-4,5-二氢-1,2,4-三嗪-3-基)哌啶-3-氨基甲酸酯(9).MS:m/z,418(20%),443(100%,M+1),465(95%,M+23)。
步骤H.(R)-2-((3-(3-氨基哌啶-1-基)-6-甲基-5-氧代-1,2,4-三嗪-4(5H)-基)甲基)-4-氟苄腈(10,化合物A)
向(R)-叔丁基1-(4-(2-氰基-5-氟苄基)-6-甲基-5-氧代-4,5-二氢-1,2,4-三嗪-3-基)哌啶-3-氨基甲酸酯(9,37mg)的二氯甲烷溶液1mL,加入三氟醋酸0.5mL,室温搅拌1h,用饱和碳酸氢钠溶液中和,二氯甲烷萃取(10mL×3),合并有机层用无水硫酸钠干燥,抽滤浓缩得粗品,经硅胶柱色谱纯化(洗脱剂:二氯甲烷/甲醇/氨水=92:6:2)得(R)-2-((3-(3-氨基哌啶-1-基)-6-甲基-5-氧代-1,2,4-三嗪-4(5H)-基)甲基)-4-氟苄腈(10)。
1H NMR(400MHz,DMSO,ppm):δ7.96(m,1H),7.36(br,1H),7.29(d,1H),5.23(s,2H),3.15(m,3H),2.72(m,2H),2.23(s,3H),1.78(d,1H),1.64(d,1H),1.47(m,1H),1.12(m,1H).MS:m/z,343(100%,M+1)。
实施例2化合物A苯甲酸盐的制备
配置95%乙醇溶液:500mL烧杯中加入228mL乙醇,加入12mL水,搅拌均匀,备用。
取2.14g苯甲酸,室温下加入10mL95%乙醇搅拌溶解,备用;向500mL反应瓶中加入化合物A精制品60g,95%乙醇120mL,搅拌,溶清,过滤,用95%乙醇18ml洗涤;使内温保持在15℃下滴加苯甲酸的乙醇溶液。滴加完毕,95%乙醇洗涤,减压干燥至恒重,得42.4g化合物A苯甲酸盐。
1HNMR(DMSO-d6,400MHz,ppm)δ7.96-7.88(m,3H),7.45-7.26(m,5H),6.80(brs,3H),5.21(q,2H),3.41(d,1H),3.11-3.08(m,2H),2.91-2.79(m,2H),2.23(s,3H),1.95-1.91(m,1H),1.78-1.74(m,1H),1.57-1.42(m,2H).MS:m/z,341(100%,M-1),343(100%,M+1).
实施例3化合物A苯甲酸盐的无定形的制备
将化合物A苯甲酸盐5.0g置于二氯甲烷溶剂25mL中,搅拌溶解,20℃~30℃浓缩至干,得气泡样物品,常温真空干燥,制备得到化合物A苯甲酸盐的无定形。
所得化合物A苯甲酸盐的无定形的X射线衍射图谱如图1所示,DSC图谱如图3所示,TG图谱如图5所示。
实施例4化合物A苯甲酸盐的无定形的制备
将化合物A苯甲酸盐5.4g置于乙腈溶剂30mL中,搅拌溶解,20℃~30℃浓缩至干,得气泡样物品,常温真空干燥,制备得到化合物A苯甲酸盐的无定形。
所得化合物A苯甲酸盐的无定形的X射线衍射图谱如图2所示,DSC图谱如图4所示。
总结:无定形的不同批次样品其X射线衍射图均无晶体的特征吸收峰,但当由本领域普通技术人员,采用相应方法得到的同一物质的无定形样品采用相同的仪器和检测方法进行检测时,其X射线衍射图曲线变化应基本一致,但特定位移对应的特定峰高可能会有略微差异,均应认为属于相同无定形的X射线衍射图。
实施例5稳定性实验
高湿实验:依据《中国药典》2010版第二部附录XIXC《原料药于药物制剂的稳定性试验指导原则》,分别精密称取实施例3与实施例4的化合物A苯甲酸盐无定形、实施例1的化合物A各5份,每份各100mg,裸露放入培养皿中,置于RH92.5%的恒温恒湿箱中(温度25±2℃),并于0天、1天、2天、13天后取样测定,结果见表1。
表1高湿实验结果(RH92.5%,25±2℃)
通过上述结果可以看出:实施例3和4所得化合物A苯甲酸盐的无定形在25℃±2℃,RH92.5%条件下放置1、2和13天后其相对含量相对于现有技术获得化合物A具有明显更好的稳定性。
实施例6药物组合物的制备
化合物A苯甲酸盐(无定形) 6.78g
糊精 84.00g
按常规方法,将上述物质混合均匀后,分1000等份分别装入普通明胶胶囊,得到1000颗胶囊。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!