一种3‑氨基甲基四氢呋喃的制备方法与流程
本发明属于化合物合成技术领域,涉及一种3-氨基甲基四氢呋喃的制备方法,具体涉及一种采用还原法制备3-氨基甲基四氢呋喃的方法。
背景技术:
3-氨基甲基四氢呋喃是医药农药等常用的中间体,现有技术中对于其制备方法的公开较少,原因在于由呋喃环的取代反应可以比较简单地衍生2位上具有取代基的四氢呋喃化合物,与之相对,合成3位上具有取代基的四氢呋喃化合物是非常困难的;目前,仅公开下述3种方法。
(一)、作为3-氨基甲基四氢呋喃的制备方法之一,已知有在氨水溶液中,氢气存在下,以四氢呋喃-3-甲醛为原料进行还原氨基化的方法。但是,该方法中,为了得到作为原料的四氢呋喃-3-甲醛,不得不使用非常昂贵的原料,所以在工业上不是有利的方法。
(二)、作为另一种制备方法,以3-羟基甲基四氢呋喃为原料,衍生为3-(四氢呋喃基)卤代甲烷或3-(四氢呋喃基)磺酸甲酯,使其与邻苯二甲酰亚胺钾反应,进行水解或肼解。但该方法中,生成大量来自于邻苯二甲酰亚胺的副产物,或使用危险性高的肼等,不是工业上有利的方法。
(三)、还有一种制备方法,以苹果酸为原料,通过高压氢化来制备2-羟基-1,4-丁二醇,并环化得到3-羟基四氢呋喃,进一步衍生为3-(四氢呋喃基)卤代烷或3-(四氢呋喃基)磺酸甲酯,通过与氰化钠反应制备得到3-氰基四氢呋喃,并将其还原为3-氨基甲基四氢呋喃。但该方法步骤较长,且使用剧毒的氰化钠,不利于工业生产。
技术实现要素:
发明目的:本发明目的在于针对现有技术的不足,提供一种原料低毒且廉价,生产过程安全、高效的3-氨基甲基四氢呋喃的制备方法。
技术方案:本发明所述的一种式(I)化合物的制备方法,将氰基乙酸乙酯与环氧乙烷在碱性介质进行环化反应,得到式(II)化合物3-氰基-γ-丁内酯,然后在还原剂的作用下将式(II)化合物3-氰基-γ-丁内酯还原得到式(III)化合物2-氨基甲基-1,4-丁二醇;最后在脱水剂的作用下将式(III)化合物脱水环合反应制备得到式(I)化合物3-氨基甲基四氢呋喃;具体方程式如下:
作为本发明的优选方案,所述的碱性介质为乙醇钠的醇溶液或叔丁醇钾的醇溶液。
作为本发明的较优选方案,所述碱性介质为乙醇钠的乙醇溶液,环化反应中氰基乙酸乙酯、乙醇钠以及环氧乙烷的摩尔比为1:1~2:1~2,进一步优化为1:1.05:1.26;环化反应中,先向乙醇钠的乙醇溶液中加入氰基乙酸乙酯在20~25℃下搅拌0.5~1.5h,再向体系中加入环氧乙烷升温至45~50℃,继续搅拌反应3~6h。
作为本发明的较优选方案,所述碱性介质为叔丁醇钾的叔丁醇溶液,环化反应中氰基乙酸乙酯、叔丁醇钾以及环氧乙烷的摩尔比为1:1~2:1~2,进一步优选为1:0.05:1.5;环化反应中,先向叔丁醇钾的叔丁醇溶液中加入氰基乙酸乙酯在20~25℃下搅拌0.5~1.5h,再向体系中加入环氧乙烷升温至45~50℃,继续搅拌反应3~6h。
作为本发明的优选方案,所述还原剂为NaBH4-NiCl2体系或红铝。
作为本发明的较优选方案,所述还原剂为NaBH4-NiCl2体系,即氯化镍-硼氢化钾体系,还原反应中氰基丁内酯、氯化镍与硼氢化钾的摩尔比为1:0.05~0.1:1~2,1:0.05:1.5;反应过程中先向氰基丁内酯的甲醇溶液中加入氯化镍,在20~25℃下搅拌0.5~1.5h;然后加入硼氢化钾升温至60~70℃,回流搅拌反应3~6h。
作为本发明的较优选方案,所述还原剂为红铝,还原反应中氰基丁内酯与红铝的摩尔比为1:1~2,进一步优化为1:1.5;反应温度为20~25℃,反应时间为6~10h。
作为本发明的优选方案,所述脱水剂为丁基三氯化锡、酸性氧化铝或浓硫酸中的一种。
作为本发明的较优选方案,所述脱水剂为丁基三氯化锡,脱水环合反应中2-氨基甲基-1,4-丁二醇和丁基三氯化锡的摩尔比为1:0.05~0.1,进一步优化为1:0.05;反应温度为140~160℃,反应时间为6~10h。
作为本发明的较优选方案,所述脱水剂为浓硫酸,脱水环合反应中2-氨基甲基-1,4-丁二醇和浓硫酸的摩尔比为1:0.01~0.05,反应温度为140~160℃,反应时间为6~10h。
有益效果:(1)本发明提供了一个较新的合成3-氨基甲基四氢呋喃的方法,该方法相对于现有的合成工艺具有如下优点:(1)原料易得,实验操作简单;(2)反应条件温和,原料利用率高,产物产率和纯度均较高,对环境污染小;(3)生产成本较低,适合工业化扩大生产。
附图说明
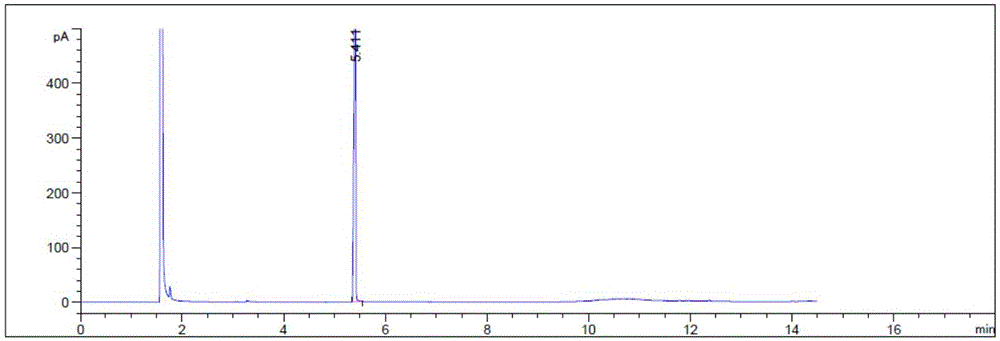
图1为本发明实施例1中3-氨基甲基四氢呋喃的气相色谱谱图;
图2为3-氨基甲基四氢呋喃标准物气相色谱谱图。
具体实施方式
下面通过具体实施例对本发明技术方案进行详细说明,但是本发明的保护范围不局限于所述实施例。
实施例1:(1)环化反应
称量7.14g(0.105mol)乙醇钠固体于250ml四口瓶中,加入50ml无水乙醇溶解,于25℃下加入氰基乙酸乙酯11.3g(0.1mol),并在25℃下搅拌1小时,出现大量白色固体。
于室温下向四口瓶中通入环氧乙烷5.54g(0.126mol),通入完毕后,升温至45~50℃搅拌4小时,白色固体消失,体系变为琥珀色澄清溶液。
对整个体系进行减压蒸处理,除去无水乙醇;对于蒸馏残余物,向蒸馏残余物中加入50ml纯净水,并用浓盐酸调节PH值为2~3;并用乙酸乙酯萃取(50ml*2),合并有机相,减压蒸除乙酸乙酯,残余物继续减压精馏;合并有机相,最终得到3-氰基-γ-丁内酯的无色澄清油状物7.8g,纯度98%,收率70%。
(2)还原反应
称量11.1g(0.1mol)3-氰基-γ-丁内酯置于250ml四口瓶中,加入50ml无水甲醇溶解,于25℃下加入氯化镍0.64g(0.005mol),并在25℃下搅拌1小时。
于25℃下分批加入硼氢化钾8.1g(0.15mol),通入完毕后,升温至65℃回流搅拌反应4小时,体系变为黑灰色悬浊液;气相检测至反应终点,然后将体系降温至25℃,向反应体系中滴加10ml纯净水,并在25℃下搅拌1小时;然后减压过滤,滤饼用10ml甲醇洗涤两次,合并有机相,减压蒸除甲醇,蒸馏残余物继续减压精馏,得到2-氨基甲基-1,4-丁二醇的无色澄清油状物10g,纯度98%,收率84%。
(3)脱水合环反应
称量10g(0.084mol)2-氨基甲基-1,4-丁二醇置于25ml四口瓶中,于室温下加入丁基三氯化锡1.18g(0.0042mol),并升温至150℃,反应8小时。气相检测至反应终点,将残余物减压精馏,得到的无色澄清油状3-氨基甲基四氢呋喃6.78g,纯度99%,收率80%。
如图1示,为本实施例中得到的3-氨基甲基四氢呋喃的气相色谱谱图,与图2所示的3-氨基甲基四氢呋喃标准物气相色谱谱图对比分析可知保留时间完全一致,由此证明本实施例中得到的是3-氨基甲基四氢呋喃。
实施例2:
(1)环化反应
称量11.78g(0.105mol)叔丁醇钾固体于250ml四口瓶中,加入50ml叔丁醇溶解,于20℃下加入氰基乙酸乙酯11.3g(0.1mol),并在20℃下搅拌1小时,出现大量白色固体。
于室温下向四口瓶中通入环氧乙烷5.54g(0.126mol),通入完毕后,升温至45~50℃搅拌3小时,白色固体消失,体系变为琥珀色澄清溶液。
对整个体系进行减压蒸处理,除去叔丁醇;对于蒸馏残余物,向蒸馏残余物中加入50ml纯净水,并用浓盐酸调节PH值为2~3;并用乙酸乙酯萃取(50ml*2),合并有机相,减压蒸除乙酸乙酯,残余物继续减压精馏;合并有机相,最终得到3-氰基-γ-丁内酯的无色澄清油状物8.1g,纯度98%,收率73%。
(2)还原反应
称量11.1g(0.1mol)3-氰基-γ-丁内酯置于250ml四口瓶中,加入50ml无水甲醇溶解,于25℃下加入氯化镍0.64g(0.005mol),并在25℃下搅拌1.5小时。
于25℃下分批加入硼氢化钾8.1g(0.15mol),通入完毕后,升温至60℃回流搅拌反应6小时,体系变为黑灰色悬浊液;气相检测至反应终点,然后将体系降温至25℃,向反应体系中滴加10ml纯净水,并在25℃下搅拌1小时;然后减压过滤,滤饼用10ml甲醇洗涤两次,合并有机相,减压蒸除甲醇,蒸馏残余物继续减压精馏,得到2-氨基甲基-1,4-丁二醇的无色澄清油状物9.88g,纯度98%,收率83%。
(3)脱水合环反应
称量10g(0.084mol)2-氨基甲基-1,4-丁二醇置于25ml四口瓶中,于室温下加入丁基三氯化锡1.18g(0.0042mol),并升温至140℃,反应10小时。气相检测至反应终点,将残余物减压精馏,得到的无色澄清油状3-氨基甲基四氢呋喃6.78g,纯度99%,收率80%。
实施例3:
(1)环化反应
称量7.14g(0.105mol)乙醇钠固体于250ml四口瓶中,加入50ml无水乙醇溶解,于25℃下加入氰基乙酸乙酯11.3g(0.1mol),并在25℃下搅拌1小时,出现大量白色固体。
于25℃下向四口瓶中通入环氧乙烷5.54g(0.126mol),通入完毕后,升温至45~50℃搅拌3小时,白色固体消失,体系变为琥珀色澄清溶液。
对整个体系进行减压蒸处理,除去无水乙醇;对于蒸馏残余物,向蒸馏残余物中加入50ml纯净水,并用浓盐酸调节PH值为2~3;并用乙酸乙酯萃取(50ml*2),合并有机相,减压蒸除乙酸乙酯,残余物继续减压精馏;合并有机相,最终得到3-氰基-γ-丁内酯的无色澄清油状物7.8g,纯度98%,收率70%。
(2)还原反应
称量11.1g(0.1mol)3-氰基-γ-丁内酯置于250ml四口瓶中,加入50ml甲苯溶解。于25℃下缓慢加入红铝的甲苯溶液(0.15mol),加入完毕后,保温25℃搅拌反应10小时;气相检测至反应终点,然后将体系降温至0℃,向反应体系中滴加10ml纯净水,并用浓盐酸调节PH至中性,在25℃下搅拌1小时;然后减压过滤,滤饼用10ml甲苯洗涤两次,合并有机相,减压蒸除甲苯,蒸馏残余物继续减压精馏,得到2-氨基甲基-1,4-丁二醇的无色澄清油状物10.47g,纯度98%,收率88%。
(3)脱水合环反应
称量10g(0.084mol)2-氨基甲基-1,4-丁二醇置于25ml四口瓶中,于25℃下加入丁基三氯化锡1.18g(0.0042mol),并升温至160℃,反应6小时。气相检测至反应终点,将残余物减压精馏,得到的无色澄清油状3-氨基甲基四氢呋喃6.85g,纯度99%,收率81%。
如上所述,尽管参照特定的优选实施例已经表示和表述了本发明,但其不得解释为对本发明自身的限制。在不脱离所附权利要求定义的本发明的精神和范围前提下,可对其在形式上和细节上作出各种变化。
- 还没有人留言评论。精彩留言会获得点赞!