C‑2位和C‑7位修饰的1‑去氧巴卡亭VI紫杉烷类化合物及其制备方法与流程
本发明涉及一种1-去氧巴卡亭VI紫杉烷类化合物及其制备方法,特别是一种C-2和C-7位修饰的1-去氧巴卡亭VI紫杉烷类化合物及其制备方法。
技术背景
紫杉醇(Paclitaxel,商品名Taxol)是从紫杉或者红豆杉树中提取的新型天然抗癌药物,具有广谱高效的抗肿瘤活性,被称为“晚期癌症的最后一道防线”。其结构如下:
近几年的研究表明紫杉醇除了具有抗癌作用外,同时还具有治疗其他疾病(如类风湿性关节炎、疟疾、老年痴呆以及中风等)的潜力。MD-2作为天然免疫识别过程的重要调控分子,有可能成为炎症性疾病治疗的“新靶点”。因此,理论上说通过阻断或拮抗MD-2与LPS的结合,可以阻断或抑制病原体信号向细胞内传递,从而达到抵御或减轻病原菌所导致的过度炎症损伤的目的。针对MD-2靶标对紫杉醇进行结构修饰,人们做了很多研究。比如:1999年Vogel等对紫杉醇的C-4,C-10,C-13进行结构改造并研究其抑制LPS刺激的TNFα的量,发现在C-2’为呋喃或C-3’为吡啶取代时活性远比紫杉醇高。2008年Jerala等对紫杉醇与紫杉特作用于MD-2进行分子对接,发现连接的强弱由疏水作用控制,因此,C-3’-N取代基改变紫杉醇类似物的亲脂性,从而影响与MD-2连接强弱。基于此,针对MD-2靶标也设计了一系列从C-2位和C-7位结构修饰的紫杉烷类似物,以1-去羟基巴卡亭VI为原料合成目标化合物,并对其抗炎症反应进行体外实验。能够得到抗炎活性较高的紫杉烷类似物。
技术实现要素:
本发明的目的之一在于提供一系列C-2位和C-7位修饰的1-去氧巴卡亭VI 紫杉烷类化合物。
本发明的目的之二在于提供该类化合物的制备方法。
为达到上述目的,本发明采用如下反应合成路线:
根据上述反应合成路线,本发明采用如下技术方案:
一类C-2位和C-7位修饰的1-去氧巴卡亭VI紫杉烷类化合物,其特征在于该类似物的结构式为:
其中:R为:苯甲酰基取代基或五元杂环取代基。
上述的苯甲酰基取代基为:所述的苯甲酰基取代基为:卤素取代的苯甲酰基、硝基取代的苯甲酰基、甲氧基取代的苯甲酰基、苯甲酰基、C1-C4烷基取代的苯甲酰基或三氟甲基取代的苯甲酰基;所述的五元杂环取代基为:噻吩甲酰基或呋喃甲酰基。
一种制备上述的C-2位和C-7位修饰的1-去氧巴卡亭VI紫杉烷类化合物的方法,其特征在于该方法的具体步骤为:
a.将1-去羟基巴卡亭VI与水合肼按1:300~400的摩尔比溶于质量百分比浓度为95%的酒精中,室温下搅拌15~18小时;调节体系的pH值为6~7,乙酸乙酯萃取,有机相经干燥,去除溶剂得粗产物;该粗产物经分离纯化,得无色透明晶体7,9,10,13-四去乙酰基-1-去氧巴卡亭VI,即化合物2,其结构式为:
b.将步骤a所得化合物2和2,2-二甲氧基丙烷按1:2~5的摩尔比溶于二氯甲烷溶剂中,再加入催化量的蒙脱土K10,在30~50℃下搅拌至反应完全;除去催化剂和溶剂后,该粗产物经分离纯化,得白色固体7,9,10,13-四去乙酰基-9,10-O-异亚丙基-1-去氧巴卡亭VI,即化合物3,其结构式为:
c.将步骤b所得化合物3和有机碱三甲基苄基氢氧化铵按1:8的摩尔比溶于二氯甲烷中(5~8mL),在室温条件下反应6~8小时;在水浴条件下用0.1N盐酸中和至PH=6~7,二氯甲烷萃取,有机相用饱和食盐水洗涤,无水硫酸镁干燥,除去二氯甲烷能得到4,7,9,10,13-五去乙酰基-2-去苯甲酰基-9,10-O-异亚丙基-1-去羟基巴卡亭Ⅵ,即化合物4,其结构式为:
d.将步骤c所得化合物4、4-二甲氨基吡啶和乙酸酐按1:1:3的摩尔比溶解在四氢呋喃中,冰水浴下搅拌反应0.5~1小时;后加入饱和碳酸氢钠水溶液,去除溶剂,乙酸乙酯萃取,去离子水洗涤,无水硫酸镁干燥,除去乙酸乙酯得粗产物,该粗产物经分离提纯得到白色固体产物4,7,9,10-四去乙酰基-2-去苯甲酰基-2-乙酰基-9,10-O-异亚丙基-1-去羟基巴卡亭Ⅵ,即化合物5,其结构式为:
e.将步骤d所得化合物5、4-二甲氨基吡啶和间位取代苯甲酸按1:1:3的摩尔比溶解在甲苯中,室温搅拌反应11~13小时;后加入饱和碳酸氢钠水溶液,乙酸乙酯萃取,去离子水洗涤,有机相用无水硫酸镁干燥,除去乙酸乙酯得粗产物,该粗产物经分离提纯得到白色固体产物4,7,9,10-四去乙酰基-2-去苯甲酰基-2-乙酰基-7-取代-9,10-O-异亚丙基-1-去羟基巴卡亭Ⅵ,即化合物6,其结构式为:
所述的间位取代苯甲酸的结构式为:
f.将步骤e所得化合物6、4-二甲氨基吡啶和乙酸酐按1:10:20的摩尔比溶解在甲苯中,室温搅拌反应15~17小时;加入饱和碳酸氢钠水溶液,乙酸乙酯萃取,去离子水洗涤,有机相用无水硫酸镁干燥,除去乙酸乙酯得粗产物,该粗产物经分离提纯得到白色固体产物7,9,10-三去乙酰基-2-去苯甲酰基-2-乙酰基-7-取代-9,10-O-异亚丙基-1-去羟基巴卡亭Ⅵ,即化合物7,其结构式为:
上述的C-2位和C-7位改造的1-去羟基巴卡亭VI紫杉烷类似物在制备抗炎药物中的应用。
本发明的1-去羟基巴卡亭VI紫杉烷类似物在C-2位将苯氧酰基改造为乙酰氧基并且在C-7位引入各种取代基,保留紫杉烷类的环骨架和必要的官能团,丰富了该类化合物,通过初筛实验,稳定性实验,浓度梯度实验等相关数据表明部分化合物在抗炎方面比紫杉醇更好,同时也为这类化合物的活性构效关系的研究提供了宝贵的借鉴。
附图说明
图1为不同药物浓度作用后LPS刺激细胞中TNF-α的表达;
图2为本发明产物7c和紫杉醇的半数致死量;
具体实施方式
下面结合具体实施例对本发明作进一步阐述,但不限制本发明。
实施例1:化合物7-间氟苯甲酰基-1-去羟基紫杉烷类似物(7a)的制法:
A.1-去羟基巴卡亭VI(化合物1)(419mg,0.6mmol)溶于20mL95%酒精中,加入10mL水合肼,室温下搅拌15小时。用0.2N稀盐酸中和,乙酸乙酯萃取,有机相用饱和食盐水洗三次,无水硫酸钠干燥,减压蒸除溶剂。粗产物用甲醇和正己烷重结晶,得无色透明晶体7,9,10,13-四去乙酰基-1-去氧巴卡亭VI(化合物2)为227mg,产率为87%。
B.化合物2(239mg,0.5mmol)溶于18mL二氯甲烷和1.5mL甲醇,完全溶解后再加入2,2-二甲氧基丙烷(0.4mL,2.0mmol),搅拌均匀,再加入蒙脱土K10为24mg,室温下搅拌0.5小时。减压蒸馏除去低沸点的溶剂,较多的乙酸乙酯溶解后,抽滤除去蒙脱土K10固体粉末,滤液水洗数次,无水硫酸钠干燥,减压蒸馏除去溶剂。粗产物用乙酸乙酯重结晶,得7,9,10,13-四去乙酰基-9,10-O-异亚丙基-1-去氧巴卡亭VI(化合物3)为236mg,产率为92%。
C.化合物3(200mg,0.35mmol)溶于5mL二氯甲烷,加入8个当量的有机碱三甲基苄基氢氧化铵(Triton B,40%在乙醇溶液中)(1.2mL,2.8mmol)。在室温条件下反应7个小时。在水浴条件下用0.1N盐酸中和至PH=6-7,二氯甲烷萃取3次,有机相用饱和食盐水洗涤,无水硫酸镁干燥,除去二氯甲烷能得到4,7,9,10,13-五去乙酰基-2-去苯甲酰基-9,10-O-异亚丙基-1-去羟基巴卡亭Ⅵ120mg(化合物4),产率为60%。
D.化合物4(51mg,0.10mmol)溶解在四氢呋喃(6mL)中,然后依次加入4-二甲氨基吡啶(122mg,0.10mmol)和乙酸酐(100mg,0.30mmol),0℃搅拌反应。半小时后加入饱和碳酸氢钠水溶液(2mL)。旋出四氢呋喃,然后加入乙酸乙酯(30mL),去离子水洗涤(3×15mL),水相再用乙酸乙酯(20mL)萃取两次。合并有机相,无水硫酸镁干燥,抽滤除去硫酸镁,旋转除去乙酸乙酯,粗产物制备硅胶板分离(展开剂:V乙酸乙酯/V石油醚=1/3)得到白色固体产物4,7,9,10-四去乙酰基-2-去苯甲酰基-2-乙酰基-9,10-O-异亚丙基-1-去羟基巴卡亭Ⅵ(化合物5)为40mg,产率为54%。
E.化合物5(51mg,0.10mmol)溶解在甲苯(6mL)中,然后依次加入4-二甲氨基吡啶(122mg,0.10mmol)和间氟苯甲酸(200mg,0.30mmol),室温搅拌反应。十二小时后加入饱和碳酸氢钠水溶液(2mL)。然后加入乙酸乙酯(30mL),去离子水洗涤(3×15mL),水相再用乙酸乙酯(20mL)萃取两次。合并有机相,无水硫酸镁干燥,抽滤除去硫酸镁,旋转除去乙酸乙酯,粗产物制备硅胶板分离(展开剂:V乙酸乙酯/V石油醚=1/3)得到白色固体产物4,7,9,10-四去乙酰基-2-去苯甲酰基-2-乙酰基-7-间氟苯甲酰基-9,10-O-异亚丙基-1-去羟基巴卡亭Ⅵ(化合物6a),产率为64-67%。
1HNMR(500MHz,CDCl3)δ(ppm):1.08(s,3H),1.16(s,3H),1.29(s,3H),1.55(s,3H),1.57(s,3H),1.90(s,3H),1.95(d,J=3.6Hz,1H),1.98(d,J=8.8Hz,1H),2.09(s,3H),2.11(s,3H),2.18(d,J=4.6Hz,1H),2.50(s,1H),2.61-2.70(m,2H),3.54(d,J=9.6Hz,1H),3.70(d,J=9.6Hz,1H),4.07(d,J=9.5Hz,1H),4.29-4.31(m,1H),4.75(d,J=9.5Hz,1H),5.13-5.15(m,1H),5.54-5.56(m,1H),5.71(d,J=8.3Hz,1H),7.28(s,1H),7.42-7.46(m,1H),7.70(d,J=9.3Hz,1H),7.83(d,J=7.8Hz,1H);
13C NMR(125MHz,CDCl3)δ(ppm):16.30,16.87,21.09,24.98,26.56,26.98,27.33,29.69,32.67,33.56,38.24,40.27,43.82,50.65,69.28,69.55,72.59,73.31, 75.66,79.17,81.12,82.20,106.87,125.37,129.89,129.96,133.19,133.24,137.29,138.83,161.50,164.65,169.95。
F.化合物6a(41mg,0.06mmol)溶解在甲苯(5mL)中,然后依次加入4-二甲氨基吡啶(73mg,0.6mmol)和乙酸酐(122mg,1.2mmol),室温搅拌反应。16小时后加入饱和碳酸氢钠水溶液1mL。然后加入乙酸乙酯30mL,去离子水洗涤(3×15mL),水相再用乙酸乙酯15mL萃取两次。合并有机相,无水硫酸镁干燥,抽滤除去硫酸镁,旋蒸除去乙酸乙酯,粗产物制备硅胶板分离(展开剂:V乙酸乙酯/V石油醚=1/3)得到白色固体产物7,9,10-三去乙酰基-2-去苯甲酰基-2-乙酰基-7-间氟苯甲酰基-9,10-O-异亚丙基-1-去羟基巴卡亭Ⅵ(化合物7a),产率为80-85%。
1HNMR(500MHz,CDCl3)δ(ppm):0.94(s,3H),1.27(s,6H),1.51(s,3H),1.63(s,3H),1.72(m,2H),1.82-1.87(m,1H),1.93(s,3H),2.11(s,3H),2.14(s,6H),2.17-2.19(m,1H),2.40-2.47(m,1H),2.78-2.84(m,1H),3.70(d,J=10.5Hz,1H),4.12-4.15(m,2H),4.31-4.32(m,1H),4.90(d,J=9.7Hz,1H),5.42-5.44(m,1H),5.79(t,J=7.7Hz,1H),6.01-6.05(m,1H),7.24-7.28(m,1H),7.40-7.44(m,1H),7.69-7.72(m,1H),7.82-7.84(m,1H);
13C NMR(125MHz,CDCl3)δ(ppm):15.44,16.05,21.28,22.60,24.92,25.59,26.26,26.30,26.58,26.97,29.70,31.96,33.89,35.48,38.90,40.36,44.29,48.01,70.22,70.40,70.69,70.93,75.28,80.38,82.24,90.06,106.56,125.54,129.78,134.83,139.60,163.41,164.68,169.21,170.25。
实施例2:同法可以制备化合物7-间氯苯甲酰基-1-去羟基紫杉烷类似物(7b),该化合物的结构式为:
6b:1HNMR(500MHz,CDCl3)δ(ppm):1.09(s,3H),1.16(s,3H),1.31(s,3H),1.58(s,3H),1.60(s,3H),1.91(s,3H),1.95(d,J=3.8Hz,1H),1.97-2.00(m,1H),2.07(t,J=8.1Hz,1H),2.10(s,3H),2.13(s,3H),2.19(d,J=6.0Hz,1H),2.43(s, 1H),2.61-2.71(m,2H),3.55(d,J=9.6Hz,1H),3.70(d,J=9.6Hz,1H),4.08(d,J=9.6Hz,1H),4.30-4.32(m,1H),4.75(d,J=9.6Hz,1H),5.14-5.16(m,1H),5.54-5.57(m,1H),5.72(t,J=5.0Hz,1H),7.42(t,J=7.8Hz,1H),7.55-7.57(m,1H),7.92-7.94(m,1H),8.00(t,J=1.8Hz,1H);
6b:13C NMR(125MHz,CDCl3)δ(ppm):16.37,16.89,21.10,24.94,24.97,25.61,26.60,27.00,27.36,29.70,32.66,33.60,33.95,38.23,40.30,43.81,50.68,53.43,69.34,73.33,75.67,79.16,81.15,82.19,106.91,127.79,129.71,132.80,134.40,137.35,138.82,164.93,169.93;
7b:1HNMR(500MHz,CDCl3)δ(ppm):0.95(s,3H),1.27(s,6H),1.28(s,3H),1.51(s,3H),1.63(s,3H),1.70-1.74(m,2H),1.82-1.87(m,1H),1.93(s,3H),2.11(s,3H),2.14(s,3H),2.19(d,J=11.3Hz,1H),2.40-2.47(m,1H),2.61(d,J=4.9Hz,1H),2.79-2.84(m,1H),3.70(d,J=10.5Hz,1H),4.11-4.15(m,1H),4.31-4.32(m,1H),4.90(d,J=9.7Hz,1H),5.41-5.44(m,1H),5.79(t,J=7.7Hz,1H),6.03(t,J=8.3Hz,1H),7.39(t,J=7.9Hz,1H),7.52-7.54(m,1H),7.92(d,J=7.8Hz,1H),8.00(s,1H);
7b:13C NMR(125MHz,CDCl3)δ(ppm):15.18,15.84,20.72,22.08,22.34,26.05,26.37,26.72,29.44,31.70,35.23,38.64,39.84,40.11,44.03,47.73,69.95,70.22,70.43,70.68,74.76,75.03,78.63,80.11,81.99,89.80,106.30,127.70,129.28,134.57,139.34,164.06,168.95,169.98,170.26。
实施例3:同法可以制备化合物7-间溴苯甲酰基-1-去羟基紫杉烷类似物(7c),该化合物的结构式为:
6c:1H-NMR(500MHz,CDCl3)δ(ppm):0.87(s,3H),1.06(s,3H),1.32(s,3H),1.65(s,3H),1.73(s,3H),1.76(m,1H),1.96(m,1H),1.98(s,3H),2.04(s,3H),2.08(m,1H),2.11(s,3H),2.51(d,J=5.0Hz,1H),2.48(m,1H),2.66(m,2H),4.22(d,J =8.5Hz,1H),4.42(m,2H),4.81(dd,J=6.5,2.5Hz,1H),4.87(d,J=9.5Hz,1H),5.44(t,J=8.5Hz,1H),5.63(m,1H),7.32(t,J=8.0Hz,1H),7.67(d,J=8.0Hz,1H),7.97(d,J=8.0Hz,1H),8.16(s,1H);
6c:13C NMR(125MHz,CDCl3)δ(ppm):16.35,16.90,21.11,24.98,25.65,26.98,27.35,29.70,32.67,33.57,38.00,40.35,43.80,48.50,69.40,69.89 72.60,73.30,75.65,79.15,81.12,82.21,106.88,125.90,129.90,129.96,133.20,133.25,137.30,138.85,163.50,164.65,169.95;
7c:1H-NMR(500MHz,CDCl3)δ(ppm):0.93(s,3H),1.24(s,3H),1.26(s,3H),1.48(s,3H),1.60(s,3H),1.72(m,2H),1.90(s,3H),2.04(d,J=4.7Hz,1H),2.09(s,3H),2.11(s,6H),2.16(d,J=8.55Hz,1H),2.41(m,1H),2.58(d,J=5.15Hz,1H),2.76(m,1H),3.67(d,J=10.45Hz,1H),4.29(m,2H),4.87(d,J=9.65Hz,1H),5.40(m,1H),5.76(t,J=7.65Hz,1H),6.00(t,J=8.35Hz,1H),7.30(t,J=7.85Hz,1H),7.66(d,J=8.15Hz,1H),7.94(d,J=7.65Hz,1H),8.12(s,1H);
7c:13C-NMR(125MHz,CDCl3)δ(ppm):14.11,15.22,21.44,21.84,22.78,26.77,26.89,27.13,27.10,21.42,34.89,38.66,42.64,43.05,48.11,69.56,71.20,72.13,74.54,76.67,80.67,82.14,84.30,106.43,122.44,128.67,129.93,133.04,133.93,135.88,138.77,164.66,169.55,169.89,170.88。
实施例4:同法可以制备化合物7-间硝苯甲酰基-1-去羟基紫杉烷类似物(7d),该化合物的结构式为:
6d:1HNMR(500MHz,CDCl3)δ(ppm):1.08(s,3H),1.17(s,3H),1.30(s,3H),1.58(s,6H),1.92(s,3H),1.98(t,J=5.0Hz,1H),2.03(t,J=3.6Hz,1H),2.06(t,J=3.6Hz,1H),2.11(s,3H),2.15(s,3H),2.20(d,J=5.8Hz,1H),2.46(s,1H),2.64-2.72(m,2H),3.56(d,J=9.7Hz,1H),3.72(d,J=9.2Hz,1H),4.10(d,J=9.6Hz,1H),4.31-4.33(m,1H),4.75(d,J=9.6Hz,1H),5.15-5.17(m,1H),5.59-5.61(m, 1H),5.73(d,J=8.2Hz,1H),7.70(t,J=8.0Hz,1H),8.37(d,J=7.8Hz,1H),8.45(d,J=8.2Hz,1H);
6d:13C NMR(125MHz,CDCl3)δ(ppm):16.42,16.90,21.11,24.98,25.60,26.66,26.98,27.36,29.71,32.66,33.58,33.92,38.23,40.32,43.79,50.58,69.50,69.89,72.44,73.32,75.70,79.14,81.13,82.19,106.90,127.28,129.62,132.81,135.34,138.91,163.65,169.92,169.95;
7d:1HNMR(500MHz,CDCl3)δ(ppm):0.92(s,3H),1.27(s,3H),1.28(s,3H),1.54(s,3H),1.64(s,3H),1.89-1.91(m,1H),1.94(s,3H),2.12(d,J=4.5Hz,1H),2.14(s,3H),2.15(s,3H),2.16(s,3H),2.20(d,J=9.1Hz,1H),2.42-2.49(m,1H),2.66(d,J=5.8Hz,1H),2.83-2.90(m,1H),3.72(d,J=10.5Hz,1H),4.11(d,J=10.5Hz,1H),4.16(d,J=9.6Hz,1H),4.32-4.34(m,1H),4.92(d,J=9.8Hz,1H),5.42-5.45(m,1H),5.86(t,J=7.7Hz,1H),6.05(t,J=9.1Hz,1H),7.68(t,J=8.0Hz,1H),8.37-8.39(m,1H),8.43-8.45(m,1H).8.85(t,J=1.9Hz,1H).
7d:13C NMR(125MHz,CDCl3)δ(ppm):15.41,16.10,21.30,22.62,26.33,26.70,26.95,29.71,31.94,35.64,38.89,40.45,44.28,47.93,70.20,70.76,71.19,75.32,80.42,82.29,84.25,90.00,106.51,124.66,127.18,129.48,132.94,133.50,133.90,135.56,139.67,163.75,164.65,169.95,170.52。
化合物7c抑制炎症反应体外实验
1.初筛实验:首先我们想知道化合物是否具有抑制炎症的效果,我们选用2种常见的Toll样受体的配基,它们是TLR4配基LPS和TLR2配基酵母多糖,分别用药物预处理后刺激剂刺激,这样我们就知道化合物是通过何种方式来抑制炎症。
2.稳定性实验:为了继续重复LPS的情况,观察稳定发挥抑制炎症作用的药物种类,我们重复了筛选实验,重复实验后,统计学分析(独立样本t检验)发现化合物7c比紫杉醇抑制炎症效果相比更好,是有意义的。(P<0.05)
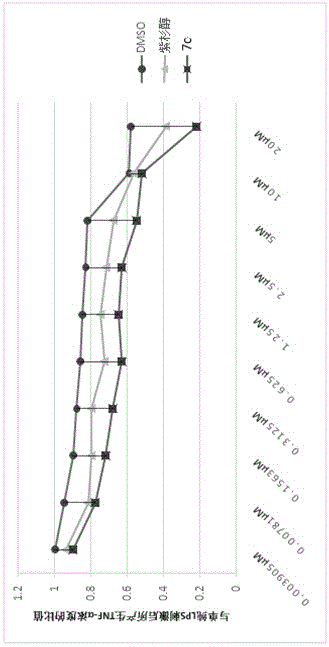
3.浓度梯度实验:为了研究化合物浓度与抑制炎症反应的的关系,我们根据不同浓度下的化合物抑制巨噬细胞分泌TNFα量,然后得出了化合物7c的抑制炎症的效果与浓度的关系图,即为图1。
从图1可以看出,随着浓度的增大,化合物抑制巨噬细胞分泌TNFα的量也随之减少。除了在浓度为10μM时,化合物7c在任意浓度下的抑炎效果比紫杉醇更好。所以,适当的C-7位修饰能够增加化合物抑制炎症反应的活性。
最后通过计算得出半数致死量,参见图2可以看出化合物7c比紫杉醇抑制炎症的效果更好。
- 还没有人留言评论。精彩留言会获得点赞!