一种N‑丁基‑2,2,6,6‑四甲基‑4‑哌啶胺的简便合成方法与流程
本发明属于化学合成技术领域,具体涉及一种N-丁基-2,2,6,6-四甲基-4-哌啶胺的简便合成的方法。
背景技术:
受阻胺光稳定剂可显著改善聚烯烃的长效光热稳定性,同时也是一类有效的抗氧剂,因此它们作为光稳定剂已广泛应用于聚乙烯、聚丙烯、聚苯乙烯、聚氯乙烯、聚醋酸乙烯、聚丙烯酸酯树脂类、聚氨酯类等树脂中,尤其它能应用于医用及食品包装材料中。
N-丁基-2,2,6,6-四甲基-4-哌啶胺是受阻胺类光稳定剂,是大分子光稳定剂的关键中间体。欧洲专利(1989年,EP302020)、中国专利(2013年,CN101522813)及文献(Catalysis Letter,2011,141(11),1703-1708.)报道了N-丁基-2,2,6,6-四甲基-4-哌啶胺的制备方法。其方法都是采用铂催化高压加氢还原胺化方法,在金属铂或氧化铂催化剂存在下,将2,2,6,6-四甲基哌啶酮和正丁胺的缩合产物在70-80℃,50bar压力下催化加氢得到N-丁基-2,2,6,6-四甲基-4-哌啶胺,收率96%。该方法的缺点是要使用高压釜和氢气,高温高压下操作不便、不安全。同时要使用昂贵的铂。
技术实现要素:
本发明的目的是克服现有技术操作不便、催化工艺成本高的缺点,简化现有技术存在的条件限制,提供一种N-丁基-2,2,6,6-四甲基-4-哌啶胺的简便合成方法。
本发明解决其技术问题所采用的技术方案是:一种N-丁基-2,2,6,6-四甲基-4-哌啶胺的简便合成方法,包括如下步骤:
1)在反应瓶内,加入2,2,6,6-四甲基哌啶酮,正丁胺和有机溶剂,搅拌溶解得混合溶液;
2)在步骤1)中混合液加入乙酸作为催化剂,搅拌混合均匀;
3)分批加入金属氢化物至步骤2)中;
4)加入水,用有机溶剂萃取;分出有机相,干燥,脱溶后减压蒸馏,收集得到N-丁基-2,2,6,6-四甲基-4-哌啶胺。
作为优选,所述的有机溶剂为二氯甲烷、二氯乙烷、乙醇、甲醇、甲苯、二甲苯中的一种或多种。
进一步地,N-丁基-2,2,6,6-四甲基-4-哌啶胺的简便合成工艺,步骤1)中2,2,6,6-四甲基哌啶酮与正丁胺的摩尔比为1.0:1.0~1.5,步骤2)中乙酸的摩尔用量是2,2,6,6-四甲基哌啶酮的5%~20%,步骤3)中所述硼氢化钠金属氢化物摩尔用量与步骤1)中2,2,6,6-四甲基哌啶酮的摩尔比例是1.0:0.25~0.75。
作为优选,步骤(2)中体系温度为0℃~60℃,反应时间5~12h。
作为优选,步骤(3)所述的反应控制体系温度在0℃~60℃温度,反应时间5~12h。
作为优选,步骤(3)所述的金属氢化物为硼氢化钠、硼氢化钾或硼氢化锂,优选为硼氢化钠。
本发明的化学还原剂还原胺化方法制备N-丁基-2,2,6,6-四甲基-4-哌啶胺的新工艺,采用乙酸为催化剂,硼氢化钠为还原剂在一定温度下还原胺化的方法,收率95.2%,避免使用高压釜和氢气,反应条件温和,经济便宜,设备简单,操作安全,容易实施。
本发明方法可以通过以下反应方程式说明:
与现有方法相比本发明具有如下有益效果:
(1)本发明所有反应条件温和,无高压高温等苛刻反应条件,无剧毒物质产生。
(2)本发明选用的硼氢化钠和催化剂乙酸价格低廉,催化效率高,收率与现有方法相当。
附图说明
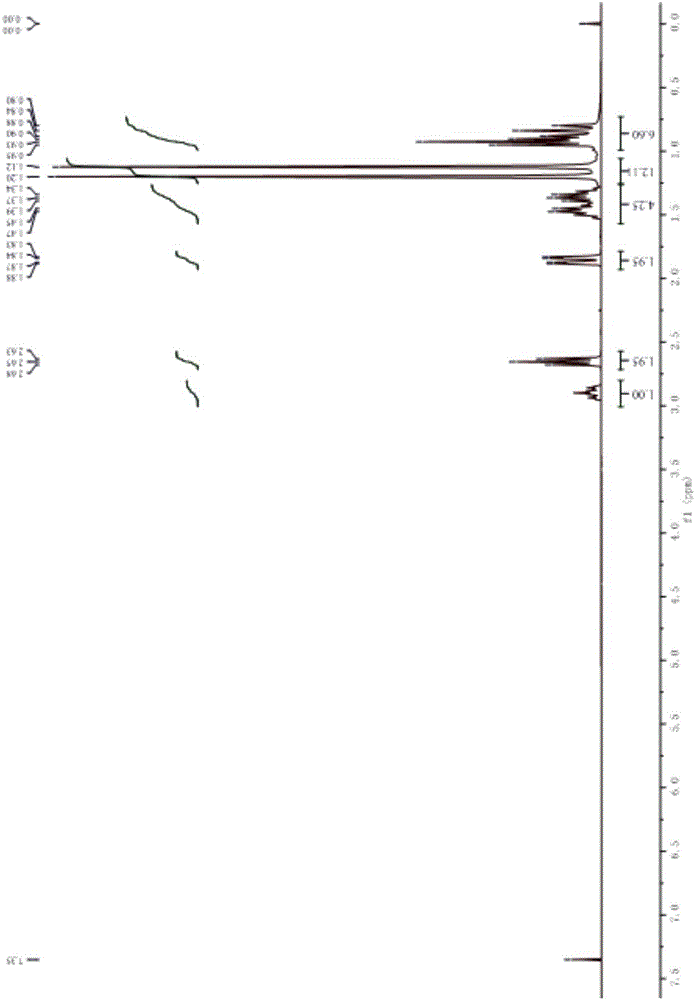
图1是产物N-丁基-2,2,6,6-四甲基-4-哌啶胺的1H-NMR图(300MHz,CDCl3)。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚明了,下面结合具体实施方式,对本发明进一步详细说明。应该理解,这些描述只是示例性的,而并非要限制本发明的范围。
实施例1
在500mL的三口烧瓶中,加入300mL甲醇溶剂,依次加入2,2,6,6-四甲基哌啶酮46.6g(0.3mol)、正丁胺24.1g(0.33mol),催化剂乙酸3.6g(0.06mol),25℃搅拌反应12h,然后分批加入硼氢化钠6.2g(0.16mol)后继续反应8h,旋蒸掉溶剂甲醇。然后加入400mL水,用乙酸乙酯(2х300mL)萃取,分出有机相,无水硫酸钠干燥,过滤后蒸去溶剂。粗产物进行减压蒸馏,收集100-102℃/5mmHg的馏分,得到淡黄色透明液体60.4g,收率为95.2%。
实施例2
在500mL的三口烧瓶中,加入300mL无水乙醇溶剂,依次加入2,2,6,6-四甲基哌啶酮46.6g(0.3mol)、正丁胺22.2g(0.3mol),催化剂乙酸3.6g(0.06mol),0℃搅拌反应5h,然后分批加入硼氢化钾4.05g(0.075mol)后继续反应8h,旋蒸掉溶剂甲醇。然后加入400mL水,用乙酸乙酯(2х300mL)萃取,分出有机相,无水硫酸钠干燥,过滤后蒸去溶剂。粗产物进行减压蒸馏,收集100-102℃/5mmHg的馏分,得到淡黄色透明液体47.2g,收率为74.2%。
实施例3
在500mL的三口烧瓶中,加入300mL二氯乙烷溶剂,依次加入2,2,6,6-四甲基哌啶酮46.6g(0.3mol)、正丁胺22.2g(0.3mol),催化剂乙酸3.6g(0.06mol),40℃搅拌反应8h,然后分批加入硼氢化钾8.6g(0.16mol)后继续反应8h。加入400mL水,分出二氯乙烷层,用乙酸乙酯(2х300mL)萃取水相,分液并合并有机相,无水硫酸钠干燥,过滤后蒸去溶剂。粗产物进行减压蒸馏,收集100-102℃/5mmHg的馏分,得到淡黄色透明液体55.8g,收率为87.5%。
实施例4
在500mL的三口烧瓶中,加入300mL无水乙醇溶剂,依次加入2,2,6,6-四甲基哌啶酮46.6g(0.3mol)、正丁胺22.2g(0.3mol),催化剂乙酸3.6g(0.015mol),60℃搅拌反应8h,然后分批加入硼氢化钠6.2g(0.16mol)后继续反应8h,旋蒸掉溶剂无水乙醇。然后加入400mL水,用乙酸乙酯(2х300mL)萃取,分出有机相,无水硫酸钠干燥,过滤后蒸去溶剂。粗产物进行减压蒸馏,收集100-102℃/5mmHg的馏分,得到淡黄色透明液体51.2g,收率为80.5%。
实施例5
在500mL的三口烧瓶中,加入300mL无水乙醇溶剂,依次加入2,2,6,6-四甲基哌啶酮46.6g(0.3mol)、正丁胺22.2g(0.3mol),催化剂乙酸3.6g(0.06mol),25℃搅拌反应12h,然后分批加入硼氢化锂3.5g(0.16mol)后继续反应5h,旋蒸掉溶剂无水乙醇。然后加入400mL水,用乙酸乙酯(2х300mL)萃取,分出有机相,无水硫酸钠干燥,过滤后蒸去溶剂。粗产物进行减压蒸馏,收集100-102℃/5mmHg的馏分,得到淡黄色透明液体57.0g,收率为89.4%。
实施例6
在500mL的三口烧瓶中,加入300mL无水乙醇溶剂,依次加入2,2,6,6-四甲基哌啶酮46.6g(0.3mol)、正丁胺22.2g(0.3mol),催化剂乙酸3.6g(0.06mol),25℃搅拌反应12h,然后分批加入硼氢化钠6.2g(0.16mol)后继续反应8h,旋蒸掉溶剂无水乙醇。然后加入400mL水,用乙酸乙酯(2х300mL)萃取,分出有机相,无水硫酸钠干燥,过滤后蒸去溶剂。粗产物进行减压蒸馏,收集100-102℃/5mmHg的馏分,得到淡黄色透明液体56.4g,收率为88.5%。
实施例7
在500mL的三口烧瓶中,加入300mL二氯甲烷溶剂,依次加入2,2,6,6-四甲基哌啶酮46.6g(0.3mol)、正丁胺22.2g(0.3mol),催化剂乙酸3.6g(0.06mol),25℃搅拌反应12h,然后分批加入硼氢化钠6.2g(0.16mol)后继续反应8h。加入400mL水,分出二氯甲烷层,用乙酸乙酯(2х300mL)萃取水相,分液并合并有机相,无水硫酸钠干燥,过滤后蒸去溶剂。粗产物进行减压蒸馏,收集100-102℃/5mmHg的馏分,得到淡黄色透明液体58.2g,收率为91.6%。
实施例8
在500mL的三口烧瓶中,加入300mL甲苯溶剂,依次加入2,2,6,6-四甲基哌啶酮46.6g(0.3mol)、正丁胺22.2g(0.3mol),催化剂乙酸3.6g(0.06mol),30℃搅拌反应12h,然后分批加入硼氢化钠6.2g(0.16mol)后继续反应8h。加入400mL水,分出甲苯层,用乙酸乙酯(2х300mL)萃取水相,分液并合并有机相,无水硫酸钠干燥,过滤后蒸去溶剂。粗产物进行减压蒸馏,收集100-102℃/5mmHg的馏分,得到淡黄色透明液体53.6g,收率为84.3%。
实施例9
在500mL的三口烧瓶中,加入300mL二甲苯溶剂,依次加入2,2,6,6-四甲基哌啶酮46.6g(0.3mol)正丁胺22.2g(0.3mol),催化剂乙酸3.6g(0.06mol),40℃搅拌反应12h,然后分批加入硼氢化钾8.6g(0.16mol)后继续反应6h。加入400mL水,分出二甲苯层,用乙酸乙酯(2х300mL)萃取水相,分液并合并有机相,无水硫酸钠干燥,过滤后蒸去溶剂。粗产物进行减压蒸馏,收集100-102℃/5mmHg的馏分,得到淡黄色透明液体54.2g,收率为85.2%。
尽管已经详细描述了本发明的实施方式,但是应该理解的是,在不偏离本发明的精神和范围的情况下,可以对本发明的实施方式做出各种改变、替换和变更。
- 还没有人留言评论。精彩留言会获得点赞!