非有机溶剂法合成赖氨匹林的方法与流程
本发明涉及赖氨匹林的制备技术领域,特别是涉及一种非有机溶剂法合成赖氨匹林的方法。
背景技术:
阿司匹林,也叫乙酰水杨酸,是应用最早、最广和最普通的解热镇痛抗炎药。该药的药理作用非常广泛,包括解热、镇痛、抗炎、治疗风湿性及类风湿性关节炎、抗血栓、减轻皮肤粘膜淋巴结综合症(川崎病)、抑制血小板聚集和预防消化道肿瘤等。而且阿司匹林还具有发挥药效迅速、药效稳定、超剂量易于诊断和处理以及很少发生过敏反应等优点。阿司匹林所具有的多种药理作用不仅为其带来了广泛的临床应用,而且也为其带来了不容忽视的不良反应。胃溃疡、胃出血、阿司匹林三联症(阿司匹林不耐受、哮喘、鼻息肉)、水杨酸反应(头痛、眩晕、耳鸣、视听力减退,甚至精神错乱、惊厥、昏迷)、肝损伤、肾损伤以及瑞氏综合症等不良反应在很大程度上限制了阿司匹林的临床应用。除此之外,阿司匹林的水溶性很差,不能制成注射剂、颗粒剂等要求药物有一定水溶性的剂型,这一点使其用药途径也受到了限制。
氨基酸是与生命活动有关的最基本的物质,是在生物体内构成蛋白质分子的基本单位,与生物的生命活动有着密切的关系。它在生物体内具有特殊的生理功能,是生物体内不可缺少的营养成分之一。在天然氨基酸中,有20种氨基酸参与蛋白质的合成。氨基酸还可作为葡萄糖的基质、氮的载体、神经递质,并与蛋白质转变、酶活性和离子通量调节有关。人类对氨基酸的需求极为广泛,其在医药领域的应用也日益增加。氨基酸衍生物类的药物在治疗肝性疾病、心血管疾病、溃疡性疾病、神经系统疾病以及炎症等方面都发挥着不可或缺的作用。此外,氨基酸衍生物还可以作为抗菌增效剂和抗生素。如今,氨基酸衍生物已被广泛用作抗肿瘤药物。这表明氨基酸衍生物的应用前景是非常广阔的。
因此,为了减轻阿司匹林的不良反应,提高其疗效,扩大其用药途径,国内外已经研制出了一系列此类药物。而赖氨匹林正是这些药物中的一种,赖氨匹林是阿司匹林和赖氨酸相结合的一种复盐,为理想的水溶性阿斯匹林盐,既有阿斯匹林的作用,又有人体必需氨基酸之一的赖氨酸的营养作用。该药不仅具有解热镇痛作用,而且还是一种良好的治疗心血管疾病的药,其抗坏血栓作用和降低动脉粥样硬化的发生率的作用优于阿司匹林。该药主要用于肌肉或静脉注射,可避免口服阿斯匹林易发生的胃肠道障碍。因此,赖氨匹林既降低了阿司匹林的不良反应,又增加了阿司匹林在水中的溶解度及给药途径。现有文献中记载的赖氨匹林的制备方法可知,现有的赖氨匹林的合成路径均需使用各种有机溶剂,这样就不可避免的会出现溶剂残留等诸多问题。
在现有的赖氨匹林合成方法及其他类似药物合成方法的基础上找到一种更合适,更简单的合成方法,以期对赖氨匹林的合成方法进行进一步的完善,并尝试将此方法运用到类似药物的合成过程中,是目前亟待解决的问题。
技术实现要素:
为了解决现有技术中存在的问题,本发明提供了一种非有机溶剂法合成赖氨匹林的方法,包括如下步骤:
S1:称取一定量的乙酰水杨酸,将所述乙酰水杨酸与浓度为4-8%的碳酸氢钠水溶液进行充分反应,制得乙酰水杨酸钠;
S2:按照摩尔比0.4-1:1的比例分别称取所述乙酰水杨酸钠和赖氨酸,将乙酰水杨酸钠加水配制成浓度为25-35%的乙酰水杨酸钠水溶液,所述赖氨酸加水配制成浓度为20-25%的赖氨酸水溶液,所述乙酰水杨酸钠水溶液与所述赖氨酸水溶液混合并发生反应,反应液用50-55℃水浴蒸发除水至有固体物质析出时停止加热,冷却至室温,在0-5℃放置18-24h使其充分析出结晶,抽滤,将滤饼干燥,即得白色粉末状的赖氨匹林。
优选地,S1中,乙酰水杨酸与碳酸氢钠的摩尔比为1:0.8-1。
优选地,S2中,乙酰水杨酸钠与赖氨酸的摩尔比为1:2。
优选地,S2中,所述乙酰水杨酸钠水溶液的浓度为30%,所述赖氨酸水溶液的浓度为22%。
优选地,S2中,所述乙酰水杨酸钠水溶液与所述赖氨酸水溶液在60℃水浴加热,回流30-40min,反应液冷却至室温,并在室温下静置24h;
反应液用50℃水浴蒸发除水至有固体物质析出时停止加热,冷却至室温,在0-5℃静置24h,充分析出结晶,抽滤,将滤饼干燥,即得白色粉末状的赖氨匹林。
优选地,S2中,将所述乙酰水杨酸钠水溶液与所述赖氨酸水溶液混合,充分搅拌使其混合均匀,于室温下静置48h;
反应液用50℃水浴蒸发除水至有固体物质析出时停止加热,冷却至室温,在0-5℃静置24h,充分析出结晶,抽滤,将滤饼干燥,即得白色粉末状的赖氨匹林。
本发明采用非有机溶剂法合成赖氨匹林,制备方法非常简单,合成过程中没有添加任何有机溶剂,有效避免了产品中有机溶剂残留,制得的赖氨匹林纯度高、产率高。
附图说明
图1为赖氨匹林标准品的红外吸收光谱图;
图2为本发明实施例1制备得到的赖氨匹林的红外吸收光谱图;
图3为本发明实施例2制备得到的赖氨匹林的红外吸收光谱图;
图4为赖氨匹林标准品的高效液相色谱图;
图5为本发明实施例1制备得到的赖氨匹林的高效液相色谱图;
图6为本发明实施例2制备得到的赖氨匹林的高效液相色谱图。
具体实施方式
为了使本领域技术人员更好地理解本发明的技术方案能予以实施,下面结合具体实施例对本发明作进一步说明,但所举实施例不作为对本发明的限定。
本发明以下实施例中所涉及的相关试剂为:碳酸氢钠(分析纯;中国派尼化学试剂厂)、乙酰水杨酸(分析纯;中国派尼化学试剂厂)、赖氨酸(分析纯;国药集团化学试剂有限公司)、甲醇(分析纯;天津市富宇精细化工有限公司)、甲醇(色谱纯;天津市富宇精细化工有限公司)、乙酸(分析纯;北京化工厂)、乙酸(色谱纯;天津市富宇精细化工有限公司)、溴化钾(KBr)(光谱纯;中国派尼化学试剂厂)、注射用赖氨匹林(产品批号:04130602;山西普德药业股份有限公司);
所涉及的相关仪器为:JA.5003A分析天平、HH-S2s数显恒温水浴锅(金坛市大地自动化仪器厂)、DHG101-1真空干燥箱(巩义市予华仪器有限责任公司)、减压抽滤真空泵(巩义市予华仪器有限责任公司)、BCD-230SDCY冰箱(青岛海尔股份有限公司)、X-5显微熔点测定仪(北京泰克仪器有限公司)、1525-高效液相色谱仪(Binary HPLC Pump;Waters)、FTIR-8201PC傅立叶变换红外分光光度计(Spectrum 100;PerkinElmer)。
一种非有机溶剂法合成赖氨匹林的方法,包括如下步骤:
S1:称取一定量的乙酰水杨酸,将所述乙酰水杨酸与浓度为4-8%的碳酸氢钠水溶液进行充分反应,制得乙酰水杨酸钠,具体的合成路线如下式(1)所示;
S2:按照摩尔比0.4-1:1的比例分别称取所述乙酰水杨酸钠和赖氨酸,将乙酰水杨酸钠加水配制成浓度为25-35%的乙酰水杨酸钠水溶液,所述赖氨酸加水配制成浓度为20-25%的赖氨酸水溶液,所述乙酰水杨酸钠水溶液与所述赖氨酸水溶液混合并发生反应,反应液用50-55℃水浴蒸发除水至有固体物质析出时停止加热,冷却至室温,在0-5℃放置18-24h使其充分析出结晶,抽滤,将滤饼干燥,即得白色粉末状的赖氨匹林,具体的合成路线如下式(2)所示。
优选地,本发明一种非有机溶剂法合成赖氨匹林的方法,具体包括以下实施例:
实施例1
(1)乙酰水杨酸钠的合成
精密称量0.2531g(3mmol)碳酸氢钠,加4ml蒸馏水,搅拌使其溶解,得到碳酸氢钠水溶液;再精密称量0.6255g(3.5mmol)乙酰水杨酸,将其加入碳酸氢钠水溶液中,搅拌使其充分反应。等到不再产生气泡的时候停止搅拌,静置,过滤,将滤液转移至蒸发皿中,于50℃水浴上蒸发除去水分。蒸干后冷却至室温,将固体物质(乙酰水杨酸钠)从蒸发皿中刮出。乙酰水杨酸钠的理论产值为0.6065g,实际产值为0.4152g,经计算产率为(0.4152g/0.6065g)×100%=68.46%。
(2)赖氨匹林的合成
精密称量0.3007g(1.5mmol)乙酰水杨酸钠,加入1ml蒸馏水,搅拌使其充分溶解,静置,过滤,配成浓度约为30%的乙酰水杨酸钠的水溶液,备用。再精密称量0.4401g(3mmol)赖氨酸,加入2ml蒸馏水,搅拌使其充分溶解,,得到赖氨酸的水溶液,备用;
将乙酰水杨酸钠和赖氨酸的水溶液加入圆底烧瓶中,60℃水浴加热,回流40min。撤去回流装置,待反应液冷却至室温,转移至10ml玻璃小瓶中,于室温下放置24h,然后用50℃水浴蒸发除水至有固体物质析出时停止加热,冷却至室温,在0-5℃的冰箱中放置24h使其充分析出结晶,抽滤,将滤饼干燥,即可得到白色粉末状的反应产物,即赖氨匹林(含有少量赖氨酸)。赖氨匹林的理论产值为0.4895g,实际产值为0.4331g,产率为(0.4331g/0.4895g)×100%=88.48%。
实施例2
(1)乙酰水杨酸钠的合成
精密称量0.2531g(3mmol)碳酸氢钠,加4ml蒸馏水,搅拌使其溶解,得到碳酸氢钠水溶液;再精密称量0.6255g(3.5mmol)乙酰水杨酸,将其加入碳酸氢钠水溶液中,搅拌使其充分反应。等到不再产生气泡的时候停止搅拌,静置,过滤,将滤液转移至蒸发皿中,于50℃水浴上蒸发除去水分。蒸干后冷却至室温,将固体物质(乙酰水杨酸钠)从蒸发皿中刮出。乙酰水杨酸钠的理论产值为0.6065g,实际产值为0.4152g,经计算产率为(0.4152g/0.6065g)×100%=68.46%。
(2)赖氨匹林的合成
精密称量0.3007g(1.5mmol)乙酰水杨酸钠,加入1ml蒸馏水,搅拌使其充分溶解,静置,过滤,配成浓度约为30%的乙酰水杨酸钠的水溶液,备用。再精密称量0.4401g(3mmol)赖氨酸,加入2ml蒸馏水,搅拌使其充分溶解,,得到赖氨酸的水溶液,备用;
将上述两种反应物的水溶液混合,搅拌使其混合均匀,转移至10ml玻璃小瓶中,于室温下放置48h,然后用50℃水浴蒸发除水至有固体物质析出时停止加热,冷却至室温,在0-5℃的冰箱中放置24h使其充分析出结晶,抽滤,将滤饼干燥,即可得到白色粉末状的反应产物,即赖氨匹林(含有少量赖氨酸)。赖氨匹林的理论产值为0.4895g,实际产值为0.2617g,产率为(0.2617g/0.4895g)×100%=53.46%。
对实施例1和实施例2所制得的赖氨匹林产品进行性能测试,具体包括熔点测定,红外鉴定和高效液相色谱鉴定,具体的测定方法及结论分别如下:
(1)熔点测定
用显微熔点仪测定实施例1和实施例2制得的赖氨匹林样品的熔点,将测得的熔点与文献中记载的赖氨匹林标准品的熔点进行比较,从而初步判断是否生成了目标产物。
用显微熔点仪测得由实施例1和实施例2制得的赖氨匹林样品的熔点分别为153.8-155.2℃、154.4-155.9℃。从文献中查得赖氨匹林的熔点为154-156℃,由此可以初步判断该产物应该为赖氨匹林。
(2)红外鉴定
用红外分光光度计测实施例1和实施例2赖氨匹林样品及标准品的红外吸收光谱,将样品及标准品的红外吸收光谱进行比较、分析,从而进一步判断是否生成了目标产物。
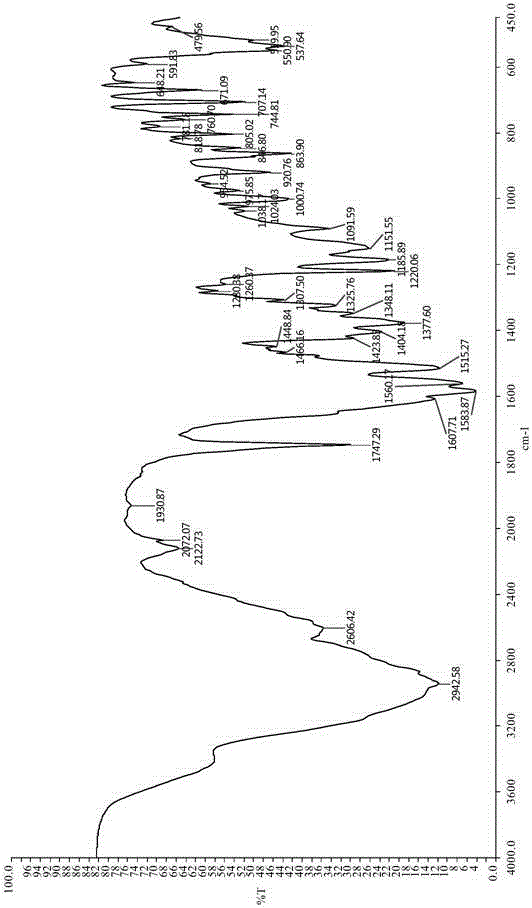
经测试,得到的赖氨匹林标准品的红外吸收光谱图,具体如图1所示,图1中苯环中碳氢键伸缩振动的吸收峰为3022.56cm-1;苯环骨架与羧基中的羰基共轭,因而其伸缩振动的吸收峰由二变四,分别为1606.40cm-1,1513.20cm-1,1583.78cm-1,1465.90cm-1;苯环中碳氢键面外弯曲振动的吸收峰为744.73cm-1,表明苯环为邻双取代结构;两个羧基中羟基伸缩振动的吸收峰为3300-2200cm-1;与苯环相连的羧基中的羰基与苯环共轭(波数减小),其伸缩振动的吸收峰为1653.14cm-1;氨基酸部分的羧基中的羰基及乙酰氧基中的羰基的伸缩振动的吸收峰为1747.20cm-1;乙酰氧基中碳氧碳键对称伸缩振动的吸收峰为1220.26cm-1,反称伸缩振动的吸收峰为1091.38cm-1;伯胺基中氮氢键对称伸缩振动的吸收峰为3489.72cm-1,反称伸缩振动的吸收峰为3403.15cm-1;甲基中碳氢键对称伸缩振动的吸收峰为2955.98cm-1,弯曲振动的吸收峰为1377.00cm-1;亚甲基中碳氢键对称伸缩振动的吸收峰为2913.25cm-1。
实施例1制备得到的赖氨匹林的红外吸收光谱图,具体如图2所示;实施例2制备得到的赖氨匹林的红外吸收光谱图,具体如图3所示;由图2-3可以看出,苯环中碳氢键伸缩振动的吸收峰为3036.72cm-1(3028.43cm-1);苯环骨架与羧基中的羰基共轭,因而其伸缩振动的吸收峰由二变四,分别为1067.71cm-1,1515.27cm-1,1583.87cm-1,1448.84cm-1(1067.87cm-1,1515.35cm-1,1583.96cm-1,1448.95cm-1);苯环中碳氢键面外弯曲振动的吸收峰为744.81cm-1(744.77cm-1),表明苯环为邻双取代结构;两个羧基中羟基伸缩振动的吸收峰为3350-2200cm-1(3350-2200cm-1);与苯环相连的羧基中的羰基与苯环共轭(波数减小),其伸缩振动的吸收峰为1657.47cm-1(1669.35cm-1);氨基酸部分的羧基中的羰基及乙酰氧基中的羰基的伸缩振动的吸收峰为1747.29cm-1(1747.27cm-1);乙酰氧基中碳氧碳键对称伸缩振动的吸收峰为1185.89cm-1(1185.68cm-1),反称伸缩振动的吸收峰为1091.59cm-1(1091.57cm-1);伯胺基中氮氢键对称伸缩振动的吸收峰为3490.12cm-1(3492.78cm-1),反称伸缩振动的吸收峰为3411.69cm-1(3403.65cm-1);甲基中碳氢键对称伸缩振动的吸收峰为2942.58cm-1(2942.59cm-1),弯曲振动的吸收峰为1377.60cm-1(1377.71cm-1);亚甲基中碳氢键对称伸缩振动的吸收峰为2904.47cm-1(2911.28cm-1),需要注意的是,上面的表述中括号内数据是图3中的,括号外数据是图2相应的。
通过比较标准品与实施例1和实施例2制得赖氨匹林样品红外吸收光谱的峰形及波数,我们可以确定标准品与样品应该是同一种物质即赖氨匹林。
(3)高效液相色谱鉴定
色谱条件:色谱柱:C18分析柱;流动相:甲醇-水-冰醋酸(40:60:1)为流动相;检测波长:276nm;流速1mL/min;柱温:25℃;进样量10μl。理论板数按赖氨匹林峰计算不低于2000。
溶液的配置:分别称取赖氨匹林标准品以及实施例1和实施例2所制得的赖氨匹林样品各0.0050g,以流动相(甲醇:水:冰醋酸=40:60:1)为溶剂配制成50ml溶液,浓度为100μg/ml。
进样测定:各物质的进样量均为10μl,记录高效液相色谱图,根据样品及标准品的保留时间、峰面积和浓度进行比较和计算。对上述三种结果进行综合分析并做出判断。
经测试,得到的赖氨匹林标准品的高效液相色谱图,具体如图4所示,实施例1制备得到的赖氨匹林的高效液相色谱图,具体如图5所示;实施例2制备得到的赖氨匹林的高效液相色谱图,具体如图6所示;
从图4-6可以看出,赖氨匹林标准品色谱图中的唯一一个峰即为赖氨匹林所出的峰,该峰的保留时间为2.979min,而实施例1和实施例2合成的赖氨匹林样品的最高峰的保留时间分别为2.975min及2.978min,经比较可知三个保留时间几乎是一致的,这表明这两份样品中均含有赖氨匹林。
用标准品的浓度及标准品与样品的峰面积可计算出各样品中赖氨匹林的含量(赖氨匹林的质量=A样C标×50×4/(A标×0.05)):实施例1赖氨匹林样品中赖氨匹林的含量为2029495×0.0001×50×4/(1995277×0.05)g=0.4069g;实施例2赖氨匹林样品中赖氨匹林的含量为721349×0.0001×50×4/(1995277×0.05)g=0.1446g;
可见,实施例1提供的合成方法和实施例2提供的合成方法都能后获得目标产物赖氨匹林,且由含量可以推断出实施例1提供的合成方法比实施例2提供的合成方法所制备的赖氨匹林的产量高。
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,其保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内,本发明的保护范围以权利要求书为准。
- 还没有人留言评论。精彩留言会获得点赞!