一种注射用盐酸氨溴索的精制方法与流程
本发明涉及化学药物纯化
技术领域:
,尤其涉及一种注射用盐酸氨溴索的精制方法。
背景技术:
:盐酸氨溴索(AmbroxolHydrochloride),化学名为反式-4-[(2-氨基3.5-二溴苄基)氨基]环已醇盐酸盐,化学结构式见下式I:盐酸氨溴索是一种祛痰药,由德国勃林格殷格翰大药厂研究开发。盐酸氨溴索注射液(商品名:沐舒坦)于上世纪80年代初在德国上市,2000年6月我国批准勃林格殷格翰大药厂生产的沐舒坦进口。盐酸氨溴索注射液临床主要用于:(1)伴有痰液分泌不正常及排痰功能不良的急性、慢性肺部疾病的治疗,如慢性支气管炎急性加重、喘息型支气管炎及支气管哮喘的祛痰治疗;(2)手术后肺部并发症的预防性治疗;(3)早产儿及新生儿的婴儿呼吸窘迫综合症(IRDS)的治疗。盐酸氨溴索的合成文献较多,如专利CN200710128121.4、CN201110445865.5、CN201210543258.7、CN201610122943.0等,但通用的合成路线为:以3,5-二溴-2-氨基苯甲醛与反式对氨基环己醇为原料,经席夫碱缩合、氢化、成盐而得,路线图如下:根据上述合成路线,盐酸氨溴索中可能引入的工艺杂质主要为反应原料3,5-二溴-2-氨基苯甲醛(杂质E)或反式对氨基环己醇、合成中间体席夫碱(杂质C)。盐酸氨溴索降解杂质与手性结构及芳胺稳定性有关,尤其是手性结构,实验证明其在高温条件下易降解成杂质B,且溶解液遇光会引起杂质A和异构体杂质D的增加,结晶清除较难。杂质A、B、C、D、E的结构如下:盐酸氨溴索杂质A、B、C、D、E清除方法较多,如专利CN201110048193.4用醇水溶液精制可有效清除杂质B、E及其他有关物质;专利CN201310045450.8用水精制能将有关物质控制在0.005%以下。但以上方法均以牺牲精制收率为代价,试验证明专利CN201110048193.4用醇水溶液精制一次的收率及专利CN201310045450.8用水精制一次的收率均不到80%,且精制母液因回收成本高,均不适合回收再精制,原料浪费严重。技术实现要素:基于
背景技术:
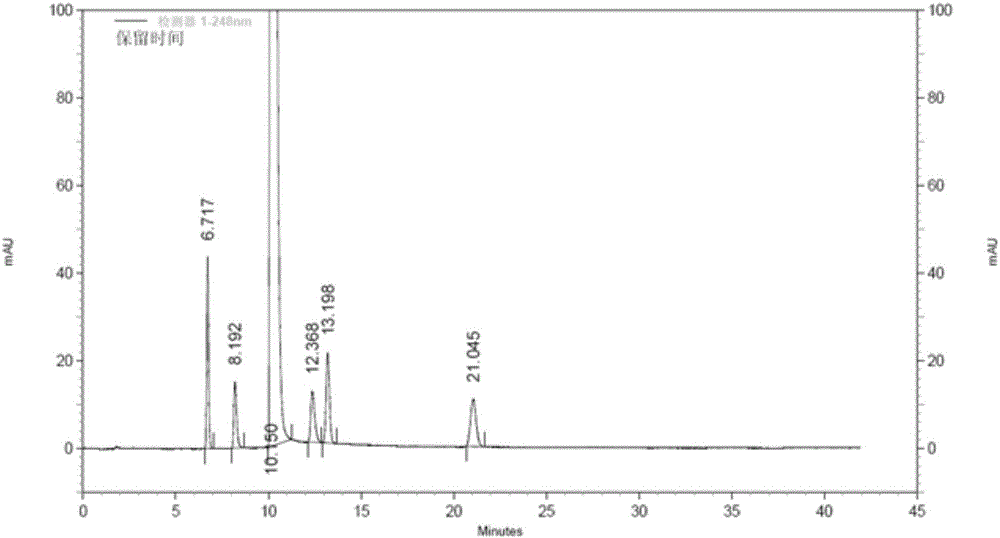
存在的技术问题,本发明提出了一种注射用盐酸氨溴索的精制方法,本发明操作简捷,成本低,产品纯度高,收率高,能有效清除杂质B,其他单个杂质的含量均可控制在0.01%以下,收率均在90%以上。本发明提出的一种注射用盐酸氨溴索的精制方法,包括如下步骤:将盐酸氨溴索粗品用盐酸水溶液加热回流溶解,过滤,取滤液重结晶,洗涤,干燥得到注射用盐酸氨溴索。优选地,将盐酸氨溴索粗品用盐酸水溶液加热回流溶解后,需趁热过滤。优选地,所述重结晶的具体步骤为:取滤液搅拌降温至72.5-73.5℃,加晶种,搅拌降温至25-30℃,然后降温至0-5℃,静置析晶,过滤取滤饼。优选地,所述重结晶的具体步骤为:取滤液搅拌降温至72.5-73.5℃,加晶种,在2-3h内搅拌降温至25-30℃,然后停止搅拌,降温至0-5℃,静置析晶2-3h,过滤取滤饼。优选地,用水洗涤,洗涤用水的温度为0-5℃。优选地,所述干燥为减压干燥。优选地,干燥的温度为75-80℃,干燥的真空度≥0.09MPa,干燥的时间为3-4h。优选地,盐酸氨溴索粗品与盐酸水溶液重量体积(g/ml)比为1:8-9。优选地,盐酸水溶液的浓度为0.01-0.08mol/L。优选地,盐酸水溶液的浓度为0.01-0.03mol/L。优选地,盐酸水溶液的浓度可以为0.02、0.03、0.04、0.05、0.06或0.07mol/L。优选地,盐酸氨溴索粗品的纯度≥98.0%。上述加入晶种量不作规定,根据具体操作确定其用量,晶种纯度≥99.99%。上述洗涤用水量不做规定,根据具体操作确定其用量。上述重结晶过程中,过滤得到的滤液为重结晶母液,可再回收,然后按照本发明精制得到注射用盐酸氨溴索。发明人在对盐酸氨溴索精制工艺条件进行筛选过程中,发现选用水、乙醇或甲醇精制,在保证收率不低于85%条件下,对含量约为0.1%的杂质的清除效果差,且杂质B特别不易清除;发明人曾尝试采用程序控温析晶,结果均不理想,最终只有增加溶剂量才显现除杂效果,但成品收率均低于80%。由此,发明人扩大了溶剂筛选范围并考察溶剂酸碱环境对杂质的影响,经试验比较,发现用浓度为0.01mol/L的盐酸水溶液对盐酸氨溴索粗品进行精制后,样品中难除性有关物质均明显降低,最大单杂的含量由0.12%降至0.003%;进一步研究发现,浓度为0.01-0.08mol/L盐酸水溶液对盐酸氨溴索杂质清除效果最好;重结晶母液去除水后所得的盐酸氨溴索粗品采用本发明精制后,最大单杂的含量小于0.005%;且选用浓度为0.01-0.03mol/L盐酸水溶液精制时,收率更高。本发明能有效清除杂质B,其他单个杂质的含量均可控制在0.01%以下,收率均在90%以上。本发明操作简捷、成本低、产品纯度高、收率高。本发明在全面总结盐酸氨溴索工艺杂质谱和降解杂质谱基础上,通过工艺合成,富集、分离提纯了杂质A、杂质B、杂质C、杂质D、杂质E,建立了全面的杂质高效液相色谱分析方法,并用该方法跟踪精制过程中杂质残留。所用高效液相色谱分析方法为:色谱柱为十八烷基硅烷键合硅胶柱(AgilentTC-C18,250mm×4.6mm,5μm);流动相为0.01mol/L磷酸氢二铵溶液(用磷酸调节pH值至7.0)-乙腈:50/50(v/v);检测波长为248nm;样品溶液的浓度为1.0mg/ml(溶剂为流动相);流速为1.0ml/min;柱温为25℃;进样量为20μl。对杂质A、B、C、D、E进行峰定位,结果见表1和图1:表1各杂质峰定位结果峰名称保留时间(min)相对保留时间杂质A6.7170.66杂质B8.1920.81盐酸氨溴索10.1501.00杂质D12.3681.22杂质E13.1981.30杂质C21.0452.07附图说明图1为盐酸氨溴索和其杂质峰定位的HPLC图谱。图2为盐酸氨溴索粗品的HPLC图谱。图3为专利CN201110048193.4精制所得盐酸氨溴索的HPLC图谱。图4为专利CN201310045450.8精制所得盐酸氨溴索的HPLC图谱。图5为本发明精制所得注射用盐酸氨溴索的HPLC图谱。具体实施方式下面,通过具体实施例对本发明的技术方案进行详细说明。实施例1一种注射用盐酸氨溴索的精制方法,包括如下步骤:将盐酸氨溴索粗品用盐酸水溶液加热回流溶解,过滤,取滤液重结晶,洗涤,干燥得到注射用盐酸氨溴索。实施例2一种注射用盐酸氨溴索的精制方法,包括如下步骤:将1kg盐酸氨溴索粗品用8L浓度为0.01mol/L盐酸水溶液加热回流溶解,趁热过滤,取滤液搅拌降温至73.5℃,加晶种1g,在2h内搅拌降温至30℃,然后停止搅拌,降温至0℃,静置析晶2h,过滤取滤饼,用200ml温度为0℃水洗涤,调节真空度为0.09MPa,调节温度至80℃,干燥3h得到0.91kg注射用盐酸氨溴索,其收率为91.0%,纯度为100%。实施例3一种注射用盐酸氨溴索的精制方法,包括如下步骤:将3kg盐酸氨溴索粗品用27L浓度为0.02mol/L盐酸水溶液加热回流溶解,趁热过滤,取滤液搅拌降温至72.5℃,加晶种1g,在3h内搅拌降温至25℃,然后停止搅拌,降温至5℃,静置析晶3h,过滤取滤饼,用1L温度为5℃水洗涤,调节真空度为0.1MPa,调节温度至75℃,干燥4h得到2.74kg注射用盐酸氨溴索,其收率为91.3%,纯度为100%。实施例4一种注射用盐酸氨溴索的精制方法,包括如下步骤:将5kg盐酸氨溴索粗品用40L浓度为0.03mol/L盐酸水溶液加热回流溶解,趁热过滤,取滤液搅拌降温至73.2℃,加晶种2g,在2.2h内搅拌降温至29℃,然后停止搅拌,降温至1℃,静置析晶3h,过滤取滤饼,用1.5L温度为1℃水洗涤,调节真空度为0.095MPa,调节温度至78℃,干燥4h得到4.58kg注射用盐酸氨溴索,其收率为91.6%,纯度为100%。实施例5一种注射用盐酸氨溴索的精制方法,包括如下步骤:将50kg盐酸氨溴索粗品用400L浓度为0.02mol/L盐酸水溶液加热回流溶解,趁热过滤,取滤液搅拌降温至72.8℃,加晶种10g,在2.8h内搅拌降温至27℃,然后停止搅拌,降温至4℃,静置析晶3h,过滤取滤饼,用10L温度为4℃水洗涤,调节真空度为0.097MPa,调节温度至76℃,干燥4h得到46.1kg注射用盐酸氨溴索,其收率为92.2%,纯度为100%。实施例6一种注射用盐酸氨溴索的精制方法,包括如下步骤:将2kg盐酸氨溴索粗品用17L浓度为0.08mol/L盐酸水溶液加热回流溶解,趁热过滤,取滤液搅拌降温至73℃,加晶种1g,在2.5h内搅拌降温至28℃,然后停止搅拌,降温至2℃,静置析晶2.5h,过滤取滤饼,用500ml温度为2℃水洗涤,调节真空度为0.093MPa,调节温度至77℃,干燥3.5h得到1.8kg注射用盐酸氨溴索,其收率为90.0%,纯度为100%。试验例1选用盐酸氨溴索粗品,分别按照专利CN201110048193.4、CN201310045450.8进行精制并检测精制后盐酸氨溴索的纯度,并与实施例2进行比较,结果见表2和图2-5,其中,图2为盐酸氨溴索粗品的HPLC图谱;图3为专利CN201110048193.4精制所得盐酸氨溴索的HPLC图谱;图4为专利CN201310045450.8精制所得盐酸氨溴索的HPLC图谱;图5为本发明精制所得注射用盐酸氨溴索的HPLC图谱。表2不同方法精制得到的盐酸氨溴索的纯度和杂质含量情况由表2和图2-5可见,采用本发明精制得到的盐酸氨溴索的纯度优于专利CN201310045450.8和CN201110048193.4;本发明能有效清除杂质B,杂质均未检出,本发明制得的盐酸氨溴索纯度在99.95%以上,单个杂质的含量均在0.01%以下。试验例2选用盐酸氨溴索粗品,分别按照专利CN201110048193.4、CN201310045450.8进行精制并与实施例2、实施例3、实施例4比较,统计各个方法的收率,结果见表3。表3不同盐酸氨溴索精制方法的收率情况由表3可见,本发明的收率均在90%以上,明显高于专利CN201310045450.8和CN201110048193.4的收率;本发明的收率高。以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本
技术领域:
的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1