酸存在下丙烯酸系化合物的SET‑LRP聚合的制作方法
本申请是中国专利申请no.201180041655.8的分案申请,该母案的申请日是2011年7月22日,发明名称为“酸存在下丙烯酸系化合物的set-lrp聚合”。
本发明涉及酸存在下丙烯酸系化合物的set-lrp聚合、以及聚合物组合物、和方法。
背景技术:
一段时间以来,已经以各种形式报道了活性自由基聚合(“lrp”),通常也称为可控自由基聚合(“crp”)。与lrp不同,常规自由基聚合常常以相对不受控的速率进行,直至发生链终止反应或单体反应物耗尽为止。一般可通过控制链终止反应物的量来控制平均分子量;但是,由于难以精确控制链终止反应可在任何单独高分子自由基链上发生的时间,所以常规自由基聚合可在聚合物分子群体中产生宽范围的聚合物链长度,从而产生高度多分散性的聚合物。当导致聚合物链终止的副反应被消除或显著减少时则发生lrp,并且通过控制扩增链增长的活性形式高分子自由基与除非转化为活性高分子自由基形式否则不能扩增链增长的非活性或休眠高分子之间活性聚合物链可逆转化的平衡来控制聚合度。因此,lrp能够通过自由基链延伸产生聚合物,其分子量容易控制并且多分散性降低。
已开发了许多不同的lrp方案,包括但不限于硝基氧(nitroxide)介导的lrp(“nmp”)、有机铋(organobismuthine)介导的lrp(“birp”)、原子转移自由基聚合(“atrp”)、单电子转移退化链转移介导的lrp(singleelectrontransferdegenerativechaintransfermediatedlrp,“set-dtlrp”)和单电子转移lrp(“set-lrp”)。set-lrp方案通常在以下可逆反应中利用铜作为活化剂来催化形成活性高分子自由基:
其中x是卤素,ln是形成配体的部分,并且r1和r2是常规基团(例如,在丙烯酸系聚合物中,r1可以是h或ch3,r2可以是羧基和羧酸盐/酯)。
文献中已报道了酸性可通过以下过程对上述活化反应产生负面影响:使形成配体的部分质子化,从而中断culnx的形成,驱使反应平衡向形成以下非活性高分子移动:
参见例如美国专利第7,332,550号和tsarevsky,n.v.&matyjaszewski,k.,chem.rev.2007,107,2270-99。这导致了进行lrp反应应避免酸性溶剂并且应对丙烯酸和甲基丙烯酸系单体上的羧基进行保护或中和。但是,丙烯酸和甲基丙烯酸的水溶性盐难以通过lrp技术聚合,认为这可由以下原因引起:单体上的阴离子羧基与金属催化剂之间形成复合物,再次中断culnx的形成并驱使反应平衡向形成非活性高分子移动。
美国专利第7,332,550号报道了使用na盐形式的四配位配体形成实体(entity)乙基-1,2-二硫代二丙烯酸(“edtdaa”)进行含甲基丙烯酸的丙烯酸系单体混合物的atrp聚合。‘550专利还报道,当将酸式edtdaa直接添加到反应混合物中时不进行atrp聚合。硫醚的反应性比其对应醚大得多,并且反应活化剂(包括升高温度、不同铜形态、自由基和乙基)可导致另外的硫代形态形成,对lrp反应产生意料之外的影响。一般而言,硫醚和硫代化合物经常伴有气味和低气味阈值。通过二硫代氨基甲酸酯和其他raft试剂物质(可由多种二硫化物前体通过atrp产生,macromolecules2009,42,3738-3742)聚合因容易产生恶臭化合物而臭名昭著。因此,期望避免二硫醚配体。
因此,需要在酸性存在下有效并且不需依赖于硫醇配体化合物的lrp技术。
技术实现要素:
根据本发明的一些示例性实施方案,发现丙烯酸系单体的set-lrp聚合出乎意料地能够耐受酸性条件。酸性源可以是溶剂(例如,含乙酸的溶剂)或在单体内容物中(例如,丙烯酸或甲基丙烯酸,其任选地与其他单体如甲基丙烯酸甲酯组合)。因此,在本发明的一个示例性实施方案中,提供了聚合物组合物,其包含衍生自反应混合物的单电子转移活性自由基聚合的丙烯酸系聚合物或共聚物,所述反应混合物包含:
(a)一种或更多种丙烯酸系单体,其包括下式的单体:
其中r1是氢或c1-3烷基并且r2是羧基或羧酸盐/酯;
(b)金属单电子转移催化剂;
(c)包含溶剂和任选的无硫配体的组分,其中所述组分或组分与单体之组合能够使所述金属单电子转移催化剂歧化;和
(d)有机卤化物引发剂;
前提是r2是羧基或所述溶剂包含含羧基的化合物,或者前提是r2是羧基和所述溶剂包含含羧基的化合物。
在b.rosen和v.percec,“single-electrontransferandsingle-electrontransferdegenerativechaintransferlivingradicalpolymerization”,chem.rev.109,5069-5119(2009)和wo2008/019100a2(本文中的附件a和b)中描述了用于进行丙烯酸系单体set-lrp聚合的技术,两篇文件的公开内容各自通过引用整体并入本文。
根据本发明的一些示例性实施方案的待聚合单体包括至少一种下式的丙烯酸系单体
其中r1是氢或c1-3并且r2是羧基或羧酸盐/酯;在一个示例性实施方案中,r1是氢。r1烷基的实例包括甲基、乙基和丙基。在一个示例性实施方案中,r1烷基是甲基。用作r2的羧酸盐/酯基团包括但不限于已被胺碱(如胺)中和成盐的羧酸盐,例如羧酸铵、羧酸钠或羧酸钾。用作r2的羧酸盐/酯基团还包括羧酸酯,其包括但不限于在烷基部分具有1-12个碳原子的(烷基)丙烯酸烷基酯、在烷基部分具有1-36个碳原子并且具有1-100个烷氧基重复单元(c2、c3、c4烷氧基)的(烷基)丙烯酸烷基烷氧基酯、具有c3-8环烷基的(烷基)丙烯酸环烷基酯、具有c7-15环烷基的(烷基)丙烯酸二环烷基酯(例如,降冰片基、异冰片基)、(烷基)丙烯酸三环酯及其组合。(烷基)丙烯酸酯单体的具体实例包括但不限于(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸丁酯、(甲基)丙烯酸-2-羟基乙酯、(甲基)丙烯酸-2-甲氧基乙酯、聚(乙二醇)甲基(丙烯酸酯)、聚(烷氧基)(烷基)丙烯酸酯和(甲基)丙烯酸丙酮缩甘油酯。
所述反应混合物可还包含本领域已知的能够通过set-lrp聚合的另一些不饱和单体。另一些单体可包括例如被卤素取代的烯烃(如氯乙烯、氟乙烯)、(甲基)丙烯酰胺(如丙烯酰胺)、n-乙烯吡咯烷酮、乙酸乙烯酯、甲基丙烯酸二甲氨基乙酯(dmaema)、二甲氨基-丙基甲基丙烯酰胺(dmapma)、α-二甲基-间异丙烯基苄基异氰酸酯(tmi)和与基团的缩合产物,如醇、烷基烷氧基(如表面活性剂)、二烷基胺、烷氧基二烷基胺、烷氧基二苄基胺、苯乙烯、丙烯腈、乙烯基苄基氯、二乙烯基苯、邻苯二甲酸二烯丙酯、季戊四醇四丙烯酸酯和二甲基丙烯酸乙二醇酯。
所述金属催化剂是通过单电子转移活化原位产生可使金属卤化物歧化的电子供体。已发现铜催化剂可用作set催化剂,其通过set活化原位形成cu(i)x。一般地,良好的set催化剂的电子供体活性相对较高。电子供体活性一般与电离电势成反比,从而可与ehomo大致协调一致。对于ehomo小于8.8ev,更特别地小于8.3ev的含铜催化剂而言,已观测到良好的set性能。这些催化剂可包含cu0(例如,粉末或金属丝形式)、和cu盐(如cu2te、cu2se、cu2s或cu2o)。与需要一定化学计量量催化剂的atrp不同,set-lrp仅需要催化量的催化剂。也就是说,已证明set-lrp催化剂利用表面介导的活化,并且聚合速率与反应混合物中催化剂的表面积成比例。例如,对于反应混合物中的等量cu(0)粉,已证明减小粉末粒径(从而增加表面积)导致聚合速率增加。虽然金属催化剂可以是任何物质形态,但是在一个示例性实施方案中,催化剂是金属丝形式,从而可提供以下益处:反应发生后容易移除和回收,并且容易测量和控制表面积(例如,通过选择金属丝的规格和长度)。
可使用多种有机卤化物引发剂中的任何一种,并且卤素的选择可以改变。在单体包含甲基丙烯酸酯的一个示例性实施方案中,引发剂中的卤素可以是cl。在单体包含丙烯酸酯和/或氯乙烯的另一个示例性实施方案中,引发剂中的卤素可以是br或i。有机卤化物引发剂的具体实例包括卤仿,如chcl3、chbr3或chi3。当chcl3用作引发剂时,可使用低水平的cucl2来调控聚合。另一些示例性有机卤化物引发剂包括α-卤代酯,如2-溴丙酸甲酯、2-氯丙酸甲酯(其可采用低水平的cucl2)、2-溴异丁酸乙酯、2-溴-2-甲基丙酸-4-甲氧基苯酚酯、双(2-溴丙酰氧基)乙烷(“bpe”)、四(2-溴丙酸)季戊四醇酯、α,ω-二(溴)-聚丁醇(大分子引发剂)。另一类示例性有机卤化物引发剂包括磺酰卤化物,例如甲苯磺酰卤化物(“toscl”)、苯氧基苯-4,4’-二磺酰氯(“pdsc”),其效率可通过避免将dmso作为溶剂和/或采用多齿脂肪族n-配体来增强。另一些示例性有机卤化物引发剂包括2-溴丙腈、苄基氯、作为大分子引发剂的苄基-聚(n-异丙基丙烯酰胺)(“pnipam”)、n-苄基-2-溴-2-甲基丙酰胺、n-苯基-2-溴-2-甲基丙酰胺、n-氯-2-吡咯烷酮和1-二硫代萘二甲酸-2-氰基丙-2-基酯。当然,本公开内容不旨在限制,并且其他有机卤化物可用作引发剂。
应选择用于set-lrp的溶剂和任选配体以有助于使cu(i)歧化为cu(0)和cu(ii)。参照以下反应方案的简化演示可更好地理解其原因:
引发(活化)步骤(kact)由从电子供体(cu(0)或其他形态)至电子受体(引发剂)的set来介导。然后,该步骤中产生的cu(i)歧化为cu(ii)和cu(0)形态。为了使set-lrp反应进行,通过活化和去活化过程在原位产生的cu(i)x必须快速歧化为cu(0)和cu(ii)x2,以具有足够量的cu(ii)x2来调控快速聚合。在一个示例性实施方案中,溶剂和任选配体,或者溶剂、任选配体与单体之组合能够将含铜金属催化剂中的铜cu(i)x歧化为cu(0)和cu(ii)x2(其中x是cl、br或i)。
通过极性或质子溶剂(包括多质子溶剂),包括但不限于水、dmf、甲醇、乙醇、dmso、乙二醇、nmp、二甲基乙酰胺、乙酸、甲酸、丙酸、草酸、丙二酸、琥珀酸、或丙三酸(carballylicacid)及其混合物,促进cu(0)的快速歧化。虽然cu(i)x易于在水或许多水性介质中歧化,但是该歧化在大多数有机溶剂,甚至是极性或质子溶剂中显著减少。增加温度可增强cu(i)x的歧化。还已表明,配体存在是使平衡常数向cu(0)歧化移动的有用手段。
已知多种配体促进金属盐(如cu(i)x)的歧化。在一个示例性实施方案中,所述配体包含n-配体基团。有用的n-配体化合物包括但不限于三(2-(二甲氨基)乙基)-胺(也称为“me6-tren”)、三(2-氨基乙基)胺(“tren”)、n,n,n′,n′,n-五甲基二亚乙基三胺(“pmdeta”)、1,1,4,7,10,10-六甲基三亚乙基四胺(“hmetea”)、聚乙烯亚胺(“pei”)、2,2′-联吡啶(“bpy”)和n-正丙基-2-吡啶基甲胺(“pr-pmi”或“haddleton配体”)。因为已表明甲基丙烯酸-2-羟基乙酯和n,n-二甲基丙烯酰胺歧化cu(i)x,所以单体的选择也可增强金属催化剂的歧化。也可使用增强歧化技术的组合。例如,bpy和pr-pmi在室温下在dmso中不表现出歧化cu(i)x,但是已表明在升高温度下介导歧化。
当配体存在时,配体的量可被调整为提供所需水平的活性。当太少配体存在时,因为表面活化减少,所以kpapp可较低,这与langmuir-hinshelwood机制一致。太多配体也可导致速率降低。一般地,当存在的配体量是反应混合物中cu(i)x量摩尔当量的约一半时,可实现最高水平的歧化,但是其他量可提供聚合速率和控制的最佳组合。
需要引发电子转移来活化set-lrp。在其最简形式中,set-lrp的活化步骤是dn+r-x→r·+(x-+dn+1),其中dn是处于氧化态n的供体d。供体(对于set-lrp在大多数情况下为cu0,在set-dtlrp中是cu0或so2-·)通过氧化将电子转移至有机卤化物r-x,导致r-x键的捕获,形成有机自由基r·和x-和dn+1。这种看起来简单的机制实际上可通过三种途径进行:
(逐步det)d″+r-x→[r-x]·-+dn+1→r·+x-+dn+1
(协同det)d″+r-x→r·+x-+dn+1
(缔合et(associativeet))d″+r-x→r·+[dn+1x]
逐步解离电子转移(det)涉及从供体至有机卤化物的单电子转移(set)以产生自由基-阴离子中间体,其随后分解以提供自由基和卤化物。协同det涉及从供体至有机卤化物的set,从而介导直接异裂而不形成自由基阴离子中间体。缔合电子转移(aet)是供体对卤化物的夺取并且不形成离子中间体。除了在通过玻璃碳电极或通过溶剂化电子的无供体还原(其中aet不适用)中之外,所有的三种过程都是有效的,并且如果不太可能则也可能是一系列混合途径。
在包含均匀有机供体或有机金属供体或不均匀金属供体的系统中,任何et过程都通过形成1∶1供体/受体遭遇络合物或前体络合物进行。该遭遇络合物的特点是最宽泛地区分电子转移的基础机制。两个金属中心之间通过属于供体和受体二者的内球配位壳层的导电桥联配体进行的电子转移正如被h.taube描述为内球电子转移(inner-sphereelectrontransfer,iset)。通过没有桥联内球配体的遭遇络合物发生的电子转移反应被称为外球电子转移(outer-sphereelectron-transfer,oset)。
虽然taubeiset中的桥联络合物经常仅是瞬时的,但是有时桥联相互作用产生稳定的混合价络合物(mixed-valencecomplex)。混合价络合物中的电子转移度可由完全定域(robin-dayi类)增加至完全离域(robin-dayiii类)。通过比较供体-受体相互作用能hda,iset和oset术语可被一般化为包括有机和有机金属供体和受体。hda值小的et反应可被认为是oset,而较大值对应iset。常规的桥联相互作用将构成大的hda值,因此,iset的两个定义是一致的。根据marcus理论通过供体和受体的自交换反应的势面可容易地模拟低hda的oset过程。
根据遭遇络合物中供体cu0或so2-·与受体r-x之间的相互作用程度,在set-lrp和set-dtlrp中的r-x断裂可潜在地通过oset或iset过程发生。在弱相互作用或无相互作用的极端情况下,将发生osetdet过程,其中供体的电子被转移至σ*r-x轨道。所得去稳定化导致逐步或协同的键裂解成r·和x-。协同解离与逐步解离很大程度上由反应环境中预期的自由基-阴离子中间体的稳定性确定。供体分子与受体分子之间的较强相互作用可导致isetdet过程。这里,虽然电子转移至σ*轨道通过供体与受体的更亲密络合物进行,但其最终导致r·和x-的异裂和扩散。如果根据taubeiset模型不发生桥联卤化物相互作用,则可根据aet进行电子转移,其中卤化物移动至供体,形成络合物和r·。
oset与iset、det与aet、以及协同与逐步之间的连续区取决于供体的结构、受体,反应介质的电子环境和温度,而这种分类可引起歧义。通过marcus-hush两态理论可容易地模拟逐步osetdet。协同osetdet需要将最初的marcus理论修改为包括通过排斥产物曲线(repulsiveproductcurve)同时断键。通过两态理论不容易模拟isetdet或aet,并且需要考虑整个供体/受体络合物。
根据参照,atrp被称为“均裂”iset过程,其中休眠形态p-x与扩增的高分子自由基之间的平衡由络合反应介导,所述反应由四个基本的起作用的反应构成:
均裂:r-x→r·+x·
电子转移:cuix→[cuiix]++e-
还原:x·+e-→x-
络合:[cuiix]++x-→cuiix2
还提出了atrp平衡常数katrp是所有的这四个基本反应的平衡常数的乘积。对与atrp有关的引发剂和休眠形态的均裂键解离的热力学进行了一系列计算研究。虽然这些研究在均裂键解离能中提供了有用趋势并且r-x键的强度明显地起活化作用,但是并不见得将atrp分解为这些基本步骤具有任何具体相关性。即使断键过程类似于均裂,也需要合并供体与受体之间的相互作用。
m.j.monteiro报道了(参见j.polym.sci.,parta:polym.chem.2008,46,146)cubr与nh2-capten的络合物介导由1-溴乙基苯(1-beb)引发的苯乙烯的自由基聚合。nh2-capten是高分子二环配体,并且在其与cui/ii的络合物中,金属应存在于配体腔的中心。在另一些金属笼(metal-caged)络合物中,因为配位点的不稳定性受限和配体的空间需求,已发现电子转移反应是外球。相似地,不太可能在cubr/nh2-capten与苯乙烯的休眠溴代链端之间形成内球遭遇络合物,因此其介导的任何活化都将通过oset发生。已报道cubr/nh2-capten在60℃下介导苯乙烯的lrp,而cubr/bpy并不如此。在100℃下,与cubr/bpy相比,cubr/nh2-capten介导的自由基聚合明显加快。因为完全密封的cubr/nh2-capten不能介导显著的去活化,所以聚合并不活跃。因此,cubr/nh2-capten被作为目标用作用于α,ω-(二溴)psty多嵌段偶联的活化剂。尽管cubr/nh2-capten显著加速苯乙烯的自由基聚合速率,但是在甲苯中仍未实现多嵌段偶联。但是,在已知介导电子转移的溶剂dmso中,可实现有效的多嵌段偶联。明显的是,用cuibr活化不需要是iset,并且如果通过oset过程不能实现较高活化速率也至少是相当的。
先前已讨论了dmso在加速cuibr/pr-pmi介导的lrp中的作用。提出了溶剂的极性增强使过渡态的电荷分离稳定并且诱导亚铜卤化物催化剂的更大分离,产生反应性可能更大的cu+/l活性形态。此外,认识到dmso可与cuii络合增强活化过程,或者与cui络合而改变其反应性。在set-lrp,或许是atrp的环境中,如果其通过电子转移过程确实进行了一定程度,则电子转移的速率ket可通过以下marcus方程模拟:
(1)
在这里,hab是供体-受体耦合常数,也称为hda;λ是溶剂重组能;h是planck常数;kb是boltzmann常数;并且t是温度。λ可根据λ=λ内+λ外(λ=λin+λout)分解。溶剂化可以三个基本方式影响过程。第一,因为λ外≈(1/n2-1/εr),其中n是折射率并且εr是相对介电常数,所以溶剂可影响外球重组能λ外。溶剂对λ内的该作用对于大多数关注的溶剂并不显著。第二,产物或反应物的选择性溶剂化可影响电子转移过程的δgo。第三,电荷转移络合物自身的相对稳定化可受溶剂化影响。在自由基夺取过程中,如atrp中的“均裂”iset过程所表明的,在过渡态/供体-受体络合物中的电荷形成有限,并且因为离开的卤化物被金属中心(在atrp中为cuⅱ)捕获,所以预计卤化物的选择性溶剂化并不显著。但是,通过配位溶剂(如dmso)使cuii选择性溶剂化可加速该介质中的atrp。但是,在不选择性溶剂化cuii的另一些极性溶剂中,增加溶剂极性或反应混合物极性导致反应kpapp降低。
set-lrp一般在极性溶剂混合物中进行,因为它们一般更适合于介导歧化。已证明,将h2o添加到有机溶剂中导致kpapp的线性增加和控制的一般增加。速率增加和改善控制均可部分归因于cuix在水性介质中的改善歧化。但是,还明显的是,溶剂自身的极性有重要作用。set-lrp外球电子转移过程中的过渡态应是电子供体cu0与电子受体引发剂或休眠扩增形态之间的供体-受体电荷转移(ct)络合物。稳定电荷转移络合物的溶剂还应增强set-lrp的速率。dmso是极性非常强的有机溶剂,所以它是用于set介导的有机反应和用于set-lrp的最佳溶剂之一绝非偶然。dimroth-reichardt参数(etn)是基于吡啶
由set-lrp中dimroth-reichardt常数高的溶剂提供的速率增强似乎是其稳定et过渡态中亚铜阳离子与通过det过程衍生的卤化物阴离子电荷分离的能力的组合。机械研究已表明关于双极性非质子性介质中苄基卤化物对cu0氧化溶解的det机制。在他们的研究中,虽然反应速率与dimroth-reichardt参数不相关,但是dmso仍然是最佳溶剂。在他们的情况下,在n-配体不存在时也发生cu0→cui氧化还原过程。因此,他们发现需要用高供体数目(dnsbcl5)溶剂使原位生成的cui阳离子进行特定的溶剂化。在set-lrp中,n-配体(例如me6-tren)代替溶剂或与其共同稳定瞬时的cui和所得的cuii形态。因此,溶剂极性的作用变得更显著。
在非过渡金属催化的set-dtlrp中,提出了so2·-介导r-i中间体的det。由连二亚硫酸钠生成的so2·-因其通过自由基机制介导全氟烷基卤化物的全氟烷基化或亚磺化脱卤(sulfinatodehalogenation)的能力(推测是通过氟烷基卤化物的异裂实现)而著称。推测由so2·-介导的亚磺化脱卤通过以下过程进行:rf-x经set介导的断裂产生rf·,然后添加so2/so2-·。虽然令人信服的是该反应可通过sn2或溴夺取(bromoniumabstraction)进行,但是通过电子自旋共振(esr)和化学捕获研究已表明溶液中存在rf·。一般地,认为该过程通过det转到σ*轨道,然后进行异裂。但是,j.m.savéant已注意到在循环伏安法实验中,与通过纯oset芳香族自由基阴离子进行的相应还原相比,cf3br的so2·-还原显著加速(参见j.am.chem.soc.1990,112,786)。savéant提出密切的iset过程或br·夺取是速率增加以及稍后将so2·-称为内球供体的原因。savéant还提出了so2与br-络合是通过夺取过程使速率加速的驱动力。wakselman对该夺取机制有争议,作为主要贬低者,他引出,在使用so2·-制备三氟甲基化时,没有观测到so2br-形成(参见j.fluorinechem.1992,59,367)。不管so2·-与cf3br的反应实质是什么,在烷基卤化物的情况下由全氟烷基卤化物得到的任何结论都不太可能有根据。在全氟烷基卤化物的情况下,氟化物的吸电子能力使c-x键极化,在卤化物中产生部分正电荷。通过该部分正电荷,全氟烷基卤化物和全氟芳香族卤化物可与路易斯碱络合,并且已被用作晶体、液晶和聚合物组装中的受体。在烷基卤化物中,卤化物上的部分电荷为负,因此预计r-x与so2·-之间的遭遇络合物亲密性更小并且更不太可能参与卤化物桥。所以,即使内球遭遇络合物可接近cf3br和其他全氟烷基卤化物,也不能推断出对于烷基卤化物可以如此。此外,在set-dtlrp中,预期的电子受体是烷基碘化物。i-和反应在水性介质中进行。相对于savéant研究的极性有机介质中的br-,水性介质中i-的溶剂化增强进一步限制了通过缔合机制产生so2i-加合物的可能性。
set途径的可能性经常由反应在电子转移抑制剂如二硝基苯(dnb)存在下是否变慢来支持。在ma在甲苯/苯酚中的cu0/me6-tren催化set-lrp中,有趣的是注意到不介导任何聚合的唯一苯酚添加剂是硝基苯酚。即使在不存在苯酚的情况下,聚合也进行,但是转化率受限并且多分散性增加。虽然没有给出硝基苯酚与set-lrp不相容的解释,但是可能是硝基苯酚充当了竞争性电子受体所致。对硝基苯酚是eo(dmso)(相对于sce)为-1.26的有效电子受体。因为硝基苯酚的质子性,即使在非质子溶剂如dmso中,也进行质子辅助的不可逆还原,产生对亚硝基苯酚和多种其他进一步被还原的加合物。因为硝基苯酚的还原电势低于参与set-lrp的有机卤化物引发剂和休眠形态或者与其水平相同,所以理所当然地,它通过不可逆还原,从而消耗催化剂来抑制set过程。
因为det可能是cu0、cu2x和so2-·介导的set-lrp和set-dtlrp的机制,所以电子亲和力和异裂键解离能与活化的热力学和动力学最为相关。因此,通过dft(b3lyp)在6-31+g*的理论水平下进行了研究以计算模型引发剂和休眠卤化物的异裂键解离能(bde)。异裂bde的趋势与关于atrp计算的均裂解离能的趋势相同,这是因为所得自由基的相对稳定性在不同结构之间不同。对于有效电子供体,如cu2x、cu0或so2·-,预计涉及通过et异裂解离的机制比主要通过均裂进行的机制快得多。此外,异裂解离过程的dft表明可进行通过自由基-阴离子中间体的逐步途径。作为瞬时中间体的自由基阴离子在有机反应中并不陌生。在除ch3chcl2之外的所有情况下,计算的自由基-阴离子中间体能量低于完全解离的自由基和阴离子。在包括更有限组计算的set-lrp的第一次公开(参见percec等,j.am.chem.soc.2006,128,14156)中,该自由基-阴离子中间体被称为“自由基-阴离子簇(radical-anioncluster)”,其可被所得的cuix/l+盐笼闭。但是,当该研究扩展为包括更多种多样的引发剂和休眠形态模型化合物时,该中间体被具体称为自由基阴离子。
自由基-阴离子中间体与中性有机卤化物的相对能量为模型引发剂和休眠卤化物的电子亲和力提供了趋势。在所有情况下,电子亲和力根据i>br>cl降低。但是,自由基-阴离子中间体的稳定性根据i<br<cl降低。发现mma休眠链是比ma休眠链更有效的电子受体,并且是比vc或乙酸乙烯酯(vac)链端更好的受体。值得注意的是,发现砷磺酰卤化物对所有形态都具有最高电子亲和力,这与其增加引发速率(见上文)相符合。有机卤化物、溴化物和碘化物之间异裂bde的变化比均裂bde之间的差异显著性小得多。稍后的研究通过g3(mp2)-rad(+)从头算法评价了键解离的热力学。这个较高理论水平的研究解决了明显的一点——解离的负熵、催化剂的氧化电势和溶剂化将在一定程度上减弱异裂oset过程的益处,并可能有利于协同过程,而不有助于其中自由基阴离子至多是瞬时形态的逐步过程。预计具有高电子亲和力的有机卤化物与cu0通过电子转移机制反应。异裂bde中相对小的差异由碘代链端与氯代链端的kpapp小于数量级的差异证实。这些观测结果与其他外球异裂解离电子转移过程相符合。这些可与atrp(与其他外球过程反应性趋势的结果更为相符)kact的103-104差异中的差异形成对比。
稍后对异裂bde和电子亲和力的计算研究的重新检查揭示了三个相关的忽略之处:(1)大多数而不是所有的稳定自由基-阴离子中间体的能量略微高于中性化合物,并且在能量上显著高于解离产物,芳基卤化物的分解就属于这种情况。(2)在大多数情况下,发现r-x键距离太长而不能被认为是自由基阴离子。(3)推测的自由基-阴离子中间体的能量高于解离自由基和阴离子的唯一情况是ch3chcl2。在该情况下,r-x键距离比其他情况短得多并且更为符合自由基阴离子。
savéant还主要通过循环伏安法和计算机建模对电极催化的解离电子转移至有机卤化物进行了仔细研究,其包括观测电子转移至具有强吸电子基团的有机卤化物(包括全氟烷基卤化物、四卤化碳、卤代乙腈和α-卤代芳基及相关化合物)的显著加速。该速率加速已被归因于相对正电性有机自由基与阴离子的较大库仑引力,其通过悬挂的吸电子基团实现。这种离子-偶极相互作用通过marcus-hush关系的校正来调节活化势垒。因为稳定化术语在以上方程式1的二次吉布斯自由能部分中,所以非常小的稳定化能可具有显著作用。因此,即使在减弱离子-偶极聚集的极性介质中,小的剩余相互作用仍然导致显著的速率加速。det的该机制被称为粘性解离(stickydissociation)。应注意的是,虽然根据marcus方程在实验和理论上并且在气相从头算研究中已证实了粘性解离,但是使用一系列介电模型的相似从头算研究通常不能发现对应于阴离子-自由基对的最小值。这并不认为是理论的失败,而是常规量子力学程序中溶剂化模拟状态的失败。还应注意的是,虽然该解离形式的确产生中间体,但是断键与电子转移协同进行。
粘性解离的作用可通过势能剖面进行最好地可视化。粘性解离中离子-偶极的存在和笼状阴离子-自由基对的形成导致纯解离曲线向morse电势过渡,降低交叉点的能量(r-x/{r·+x-}的非绝热过渡态或固有的自交换能量)。在真正的自由基阴离子可早于阴离子-自由基对形成的情况下也可观测到该作用。
更新的研究提供了一些新发现(参见以上参考的b.rosen和v.percec,“single-electrontransferandsingle-electrontransferdegenerativechaintransferlivingradicalpolymerization”,chem.rev.109,5069-5119(2009),其公开内容通过引用整体并入本文。首先,几乎所有调查的set-lrp相关化合物(具有吸电子官能性的有机卤化物)中的势能剖面与关于发现进行粘性解离的其他系统所报道的势能剖面类似。特别地,在对应于库仑离子-偶极吸引力的大c-x键距离下观测到最小值。关于mma-cl示出了一个实例。在这些情况下,电荷几乎全部定位在卤化物上,而相反地,自旋主要集中在碳中心上。第二,虽然原始研究中的电子亲和力和稳定化趋势大体上重复,但是因为在不同键距离下发现的能量极小值小于先前报道的那些,所以做了一些校正。最值得注意的是,在ch2cl2异常的情况下,在较大距离发现了较低的能量粘性阴离子-自由基对,这与其他化合物一致。但是,可能的是,在ch2cl2的det中,真正的自由基-阴离子中间体可早于阴离子-自由基中间体,从而提供逐步det途径。均裂与异裂解离曲线的这种交叉允许得到待调查有机卤化物的固有自交换势垒的粗近似值。所报道的值是在相对低的理论水平下计算的未经校正的能量。但是,它不是这种重要操作的定量性质,而是暗示趋势。特别地,我们感兴趣的是,目前未通过实验研究的哪些单体和引发剂可与set-lrp相容。发现丙烯腈、甲基丙烯腈、苯乙烯、α-甲基苯乙烯、溴乙烯、氟乙烯、乙酸乙烯酯和2-氯丙烯都具有与已表明可与set-lrp相容的单体相当的电子亲和力和阴离子-自由基对稳定化能量。因此,在合适条件下预计了它们的set-lrp。
此外,发现了虽然某些有机卤化物(例如,头对头和头对尾聚(偏二氟乙烯)(pvdf))的活化比其他有机卤化物的活化明显更不利,但是它们也是可能的。已报道,cucl/me6-tren可在nmp中经聚(偏二氟乙烯)引发介导mma与氧乙烯丙烯酸酯的接枝聚合。该聚合最初被报道为atrp过程。已报道nmp与me6-tren的溶剂/配体组合介导cuix的歧化,因此更可能是set-lrp机制。此外,已报道了nmp中ma的cu0-金属丝/me6-trenset-lrp。这由与接近室温条件的相容性,纵使是均裂惰性的c-f键也如此来支持。此外,相对于4,4’-二甲基-2,2’-二吡啶基(dmdp,bpy类似物),使用me6-tren作为配体导致了巨大的速率增加。与这可归因于cuicl/me6-tren相对cuicl/dmdp的反应性增强相比,可更容易地归因于通过me6-tren更快速歧化以提供cu0而不是cui催化过程,其直接类似于以下方式:不考虑配体选择的cu0在vc聚合中提供稳定的休眠邻二卤化物的再活化,而cuix催化剂统一不能。此外,得到了这样的结论:无论出于何种目的,相对于cuicl/bpy聚合,cuicl/me6-trenmma聚合中失去了反应控制。在cuicl/me6-tren催化的由pvdf引发的mma延伸聚合中,将bpy类似物换成me6-tren作为配体增加了反应速率并且不减少分子量分布的控制。实际上,对于所报道的转化,该控制更好。在cui介导的pvdf活化中,会产生cui氟化物络合物。不清楚的是,该产物以何种程度介导去活化,从而形成休眠氯化物链端。但是,因为me6-tren中发生歧化,所以生成了cuiicl2/me6-tren去活化剂而不经过复合物络合物。因此,cu0/me6-tren可能通过增加的kact提供速率加速,同时歧化提供控制,因此是set-lrp。
附图说明
图1a-b、2a-b和3a-b描述了50℃下,0.5ml甲醇/水(95/5)中的1.00mlmma与1%乙酸(图1a-b)、2.5%乙酸(图2a-b)和25%乙酸(图3a-b)的set-lrp的结果。
图4a-b、5a-b和6a-b描述了50℃下,0.5ml不同溶剂中1.00mlmma与10%乙酸的set-lrp的结果。溶剂是甲醇/水(95/5)(图4a-b)、乙醇/水(图5a-b)和dmso(图6a-b)。
图7展示了50℃下,0.5mlacoh中1.0mlmma的set-lrp的动力学图。
图8a和8b展示了50℃下0.50ml甲醇/水(95∶5)中1.00mlmma与2.5%maa的set-lrp。
图9a中可看到增加mma的水平,并且在50℃下通过set-lrp使一批包含10%maa的1.00mlmma在0.50ml甲醇/水(95∶5)中聚合这个实验的动力学图。与之前的情况相比,该反应更慢,但是仍然遵循一级动力学,kpapp=0.004分钟1。两种单体同时并入增长链。由图9d可以看出,mma和maa二者的降低几乎完全同步发生。在ieff=50%的情况下,与单体消耗呈单调关系的分子量显著降低(图9b和9c)。
图10a-b描述了50℃下,0.50ml甲醇/水(95/5)中1.00mlmma与25%maa的聚合([mma]0/[maa]0/toscl]0/me6-tren]0=200/20/1/0.1)。
图11a-b描述了50℃下,0.50ml甲醇/水(95/5)中1.00mlmma与10%maa的聚合([mma]0/[maa]0/toscl]0/me6-tren]0=200/20/1/0.1)。
具体实施方式
通过以下非限制性实施例进一步描述本发明。
实施例
实施例1-7
根据以下反应方案,使用toscl作为自由基引发剂并用不同浓度的丙烯酸通过set-lrp进行了甲基丙烯酸甲酯(mma)与乙酸(aa)在质子性介质中的共聚:
制备单体(mma,1.00ml,9.4mmol)、乙酸(54μl,0.94mmol)溶剂(0.50ml)、toscl(9.0mg,0.047mmol)、cu(0)催化剂(12.5cm的20号金属丝,其缠绕涂有聚四氟乙烯的搅拌棒)和配体(me6-tren,1.3μl,0.004mmol)的溶液并转移到25mlschlenk管中。然后使反应混合物通过六次冷冻解冻泵循环(freeze-pump-thawcycle)脱气,放到在所需温度恒温的油浴中并搅拌。用氮气净化管的侧壁,然后在预定时间打开管以通过气密注射器移出样品。将样品溶解于cdcl3,通过1hnmr光谱测量转化率。通过gpc用pmma标准物测定mn和mw/mn值。将聚合混合物溶解于5mlch2cl2,然后通过小的碱性al2o3色谱柱以移除任何残余的新生cu(0)催化剂和cu(ii)去活化剂。将所得溶液在50ml甲醇中沉淀两次并搅拌。通过过滤除去甲醇,并将最终的无色聚合物在真空下干燥至达到恒重。
cu丝(来自fisher的20号)、甲醇(meoh)、乙醇(etoh)(fisher,有acs执照,99.9%)和乙酸(98%,aldrich)直接使用。对甲苯磺酰氯(99%,aldrich)用己烷重结晶两次。二甲亚砜(dmso)(99.9%,acros)使用前在减压下蒸馏。甲基丙烯酸甲酯(mma)购买自aldrich并且通过碱性氧化铝以除去抑制剂。六甲基化三(2-氨乙基)胺(me6-tren)如inorg.chem.1966,5,41-44所述进行合成。
20℃下在cdcl3中以四甲基硅烷(tms)作为内标物在brukerdrx500nmr仪上记录500mhz1hnmr谱。在perkin-elmer系列10的高效液相色谱仪上进行聚合物样品的凝胶渗透色谱(gpc)分析,所述色谱仪配备有超过(40℃)的lc-100柱、nelson分析990系列集成数据站、perkin-elmer785a紫外可见光检测器(254nm)、varianstar4090折射率(ri)检测器和两根am凝胶(
实施例1-3
图1a-b、2a-b和3a-b描述了50℃下,0.5ml甲醇/水(95/5)中的1.00mlmma与1%乙酸(图1a-b)、2.5%乙酸(图2a-b)和25%乙酸(图3a-b)的set-lrp的结果。[mma]0/[toscl]0/me6-tren]0之比是200/1/0.1。制备聚合溶液并相对于toscl添加2、5、20和50当量的acoh(1、2.5、10和25%的单体浓度)。在所有的这些情况下,在几小时反应时间内都得到了非常高甚至完全的转化率。聚合遵循一级动力学,并且聚合的表观速率常数kpapp随[acoh]0以一定程度增加并且在0.008至0.012分钟-1变化。不清楚这是否是一个趋势,因为发现对于相同条件下但不存在乙酸的mmaset-lrp,kpapp=0.010分钟1。(参见fleischmann,s.&percec.,v.,j.polym.sci.parta:polym.chem.,48,2010,2236-2242)。
一般地,分子量演变(molecularweightevolution)遵循set-lrp的共同原则。链增长随转化率线性进行并且计算分子量与实验分子量之间有着良好的一致性。acoh存在下的ieff似乎显著降低并且发现值为70%-80%。set-lrp过程的另一个事实是,多分散指数随转化率降低,并最终实现相当窄的分子量分布——mw/mn~1.3。在一些实施例中,最后两个数据点表明mw/mn值的增加。不能排除这是因为反应混合物在该阶段的高粘度所致。尽管如此,25%乙酸的实验尤其证明了set-lrp过程似乎相当耐受酸性介质。图1-3示出反应进行而不经过诱导期并且在240分钟内达到非常高的转化率(>95%),维持一级动力学。
实施例4-6
也没有光谱学证据证明acoh的主要淬灭是通过用甲醇原位酯化进行。这与在其他溶剂系统中进行的实验完全一致。图4a-b、5a-b和6a-b描述了50℃下,0.5ml不同溶剂中1.00mlmma与10%乙酸的set-lrp的结果。溶剂是甲醇/水(95/5)(图4a-b)、乙醇/水(图5a-b)和dmso(图6a-b)。[mma]0/[toscl]0/me6-tren]0之比是200/1/0.1。图4-6示出50℃下在甲醇、乙醇和dmso中mma与10%乙酸内容物进行的set-lrp。dmso中的反应遵循一级动力学(kpapp=0.019分钟1)并且在180分钟内达到完全转化。分子量随转化率单调增加并且mw/mn保持相当低,为1.3。meoh(kpapp=0.012分钟1)与etoh(kpapp=0.008分钟1)中的比较表现出速率的差异,这是关于中性介质中甲基丙烯酸酯set-lrp的早期研究中也已观测到的事实。在任何情况下,对分子量演变的控制在两种情况下都非常高。
实施例7
本实施例解决了set-lrp是否可在乙酸作为溶剂时进行的问题。解决的主要点是acoh是否不可逆地破坏cu(ii)/me6-tren络合物并且是否有利于cu(i)形态的歧化。在图7中,展示了50℃下,0.5mlacoh中1.0mlmma的set-lrp的动力学图。[mma]0/[toscl]0/me6-tren]0之比是200/1/0.1。直接看到了两方面。第一,反应可被驱动完成。这是很重要的观察结果,因为过度终止将引起聚合过程中断。第二,单体消耗遵循一级动力学使得总自由基浓度必须保持相当恒定。
实施例8-11
根据以下反应方案,使用toscl作为自由基引发剂并用不同浓度的甲基丙烯酸通过set-lrp进行了mma与甲基丙烯酸(maa)在质子性介质中的共聚:
制备mma(1.00ml,9.4mmol)、maa(80μl,0.94mmol)溶剂(0.50ml)、toscl(9.4mg,0.047mmol)、cu(0)催化剂(12.5cm的20号金属丝,其缠绕涂有聚四氟乙烯的搅拌棒)、配体(me6-tren,1.3μl,0.004mmol)和约20mg二甲亚砜(作为nmr实验定量的内标物)的溶液并转移到25mlschlenk管中。然后使反应混合物通过六次冷冻解冻泵循环脱气,放到在所需温度恒温的油浴中并搅拌。用氮气净化管的侧壁,然后在预定时间打开管以通过气密注射器移出样品。将样品溶解于cdcl3,通过1hnmr光谱测量转化率。通过gpc用pmma标准物测定mn和mw/mn值。将聚合混合物溶解于2mlch2cl2,然后通过小的碱性al2o3色谱柱移除任何残余的新生cu(0)催化剂和cu(ii)去活化剂。将所得溶液在50ml甲醇中沉淀两次并搅拌。通过过滤除去甲醇,并将最终的无色聚合物在真空下干燥至达到恒重。
cu丝(来自fisher的20号)和甲醇(meoh)(fisher,有acs执照,99.9%)直接使用。对甲苯磺酰氯(99%,aldrich)用己烷重结晶两次。甲基丙烯酸甲酯(mma)和甲基丙烯酸购买自aldrich并且通过碱性氧化铝以除去抑制剂。六甲基化三(2-氨乙基)胺(me6-tren)如实施例1-7中m.ciampolini所述进行合成。
20℃下在cdcl3中以四甲基硅烷(tms)作为内标物在brukerdrx500nmr仪上记录500mhz1hnmr谱。在perkin-elmer系列10的高效液相色谱仪上进行聚合物样品的凝胶渗透色谱(gpc)分析,所述色谱仪配备有超过(40℃)的lc-100柱、nelson分析990系列集成数据站、perkin-elmer785a紫外可见光检测器(254nm)、varianstar4090折射率(ri)检测器和两根am凝胶(
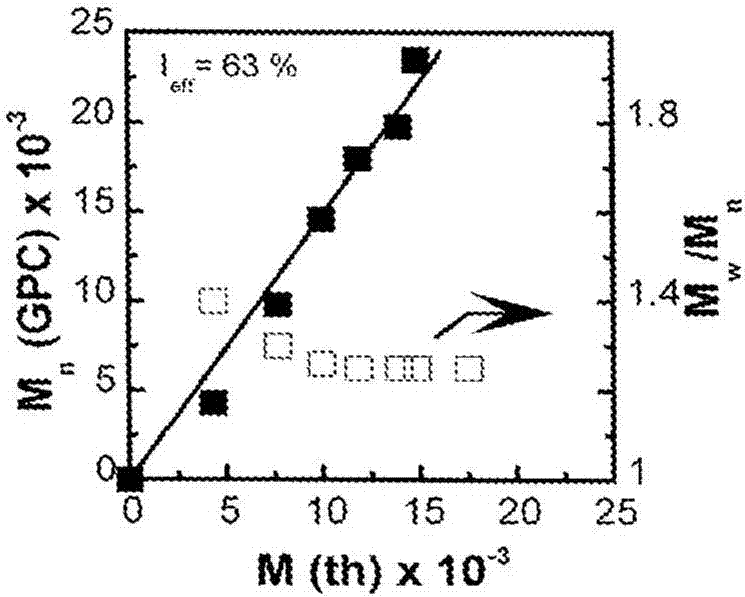
在图8a和8b中,展示了50℃下0.50ml甲醇/水(95∶5)中1.00mlmma与2.5%maa的set-lrp。[mma]0/[maa]0/toscl]0/me6-tren]0之比是200/5/1/0.1。在这个非常低的酸性单体含量下,聚合遵循一级动力学(kpapp=0.008分钟1)并且在360分钟后达到非常高的转化率。因此扩增的表观速率常数kpapp与关于不存在任何酸性(kpapp=0.010分钟1)或存在2.5%acoh(kpapp=0.009分钟1)的set-lrp的文献中所报道的范围相同。分子量随转化率线性演变,并且引发剂效率ieff=67%。mw/mn~1.3的低值表明了该过程的活性。
在下一步中,增加mma的水平,并且在50℃下通过set-lrp使一批包含10%maa的1.00mlmma在0.50ml甲醇/水(95∶5)中聚合。[mma]0/[maa]0/toscl]0/me6-tren]0之比是200/20/1/0.1。图9a中可看到这个实验的动力学图。与之前的情况相比,该反应更慢,但是仍然遵循一级动力学,kpapp=0.004分钟1。两种单体同时并入增长链。由图9d可以看出,mma和maa二者的降低几乎完全同步发生。在ieff=50%的情况下,与单体消耗呈单调关系的分子量显著降低(图9b和9c)。mw/mn值随反应进行变得更小,但是向着较高转化率略微增加。还不清楚的是,分子量分布增宽是具有真正的物理原因还是起因于取样时引入氧气的假象。与此相反的是,通过添加乙酸(与maa等摩尔)进一步增加酸性的实验揭示了一些有趣的趋势。单体消耗遵循相同的一级动力学,kpapp=0.006分钟1。此外,分子量随转化率线性增加,并且ieff=58%与之前的情况仅有略微不同。相比之下,mw/mn值等于1.3,并且在反应结束时观测到显著增加。清楚地,通过添加acoh的“额外”酸性并不进一步干扰mma的set-lrp,而是增强聚合。
图10a-b描述了50℃下,0.50ml甲醇/水(95/5)中1.00mlmma与25%maa的聚合([mma]0/[maa]0/toscl]0/me6-tren]0=200/20/1/0.1),图11a-b描述了50℃下,0.50ml甲醇/水(95/5)中1.00mlmma与10%maa的聚合([mma]0/[maa]0/toscl]0/me6-tren]0=200/20/1/0.1)。如图10a和10b所示,具有25%maa的聚合导致“活力”损失。反应在高达约50%的转化率内遵循一级动力学,并且该线性方式中的扩增速率(kpapp=0.0035分钟1)比之前的实验慢得多。超过该转化率,速率显著降低。但是,分子量随转化率线性增加,从而维持对聚合的一定控制。但是,在实施例3中,对于相同条件下在25%乙酸存在下mma的set-lrp,反应在4小时内完成。在这里,反应遵循一级动力学并且分子量随转化率线性增加。因此,反应混合物的酸性不太可能导致活化剂/去活化剂络合物的破坏,并因此不对该过程中活力的损失负责。
在本文中必须解决的一个重要问题是增长的高分子自由基形态的反应性和特性。不仅仅需要考虑mma相对maa自由基的反应性。buback等(参见macromolecules2009,42,7753-61)已证明,自由基聚合中甲基丙烯酸的聚合速率主要取决于电离度。更精确地,非电离maa的kp比完全电离的maa高出一个数量级。图11a-b所示的添加“额外”乙酸可以以使增长的maa自由基保持质子化的这种方式非常好地有助于聚合过程。聚合maa的电离度与单体显著不同,这是因为聚(甲基丙烯酸)的pka显著低于其单体的pka。
另一方面,在控制是基于休眠/活性形态的活化和去活化平衡速率的活性自由基聚合中,自由基反应性的显著差异是决定因素。set-lrp中的活化/去活化分别通过碳卤双键的异裂键断裂和卤化物可逆原子转移至以碳为中心的自由基来实现。进而,卤素在平衡休眠/活性均衡中并最终在控制聚合过程中发挥关键作用。
虽然仅根据有限数目的实施方案详细描述了本发明,但是应容易理解的是,本发明并不限于这些公开的实施方案。相反地,可将本发明修改成包括迄今为止未描述而与本发明的精神和范围相称的任何数目的变体、改变、替换或等效方案。此外,虽然已描述了本发明的多个实施方案,但是还应理解本发明的一些方面可仅包括所述实施方案中的一些。因此,本发明并不被认为是受以上描述限制。
以下内容对应于母案申请的原始权利要求书:
1.一种聚合物组合物,其包含衍生自反应混合物的单电子转移活性自由基聚合的丙烯酸系聚合物或共聚物,所述反应混合物包含:
(e)一种或更多种丙烯酸系单体,其包括下式的单体:
其中r1是氢或c1-3烷基并且r2是羧基或羧酸盐/酯;
(f)金属单电子转移催化剂;
(g)包含溶剂和任选的无硫配体的组分,其中所述组分或组分与单体之组合能够使所述金属单电子转移催化剂歧化;和
(h)有机卤化物引发剂;
前提是r2是羧基或所述溶剂包含含羧基的化合物;或者前提是r2是羧基并且所述溶剂包含含羧基的化合物。
2.根据项1所述的聚合物组合物,其中所述金属单电子转移催化剂包含铜,并且所述组分或组分与单体之组合能够使所述金属催化剂cu(i)x歧化为cu(0)和cu(ii)x2,其中x是cl、br或i。
3.根据项2所述的聚合物组合物,其中所述配体包含n-配体基团。
4.根据项1所述的聚合物组合物,其中所述配体包含n-配体基团。
5.一种制备丙烯酸系聚合物或共聚物的方法,其包括进行反应混合物的单电子转移活性自由基聚合,所述反应混合物包含:
(e)一种或更多种丙烯酸系单体,其包括下式的单体:
其中r1是氢或c1-3烷基并且r2是羧基或羧酸盐/酯;
(f)金属单电子转移催化剂;
(g)包含溶剂和任选的无硫配体的组分,其中所述组分或组分与单体之组合能够使所述金属单电子转移催化剂歧化;和
(h)有机卤化物引发剂;
前提是r2是羧基或所述溶剂包含含羧基的化合物,或者前提是r2是羧基和所述溶剂包含含羧基的化合物。
6.根据项5所述的方法,其中所述金属单电子转移催化剂包含铜,并且所述组分或组分与单体之组合能够使所述金属催化剂cu(i)x歧化为cu(0)和cu(ii)x2,其中x是cl、br或i。
7.根据项6所述的方法,其中所述配体包含n-配体基团。
8.根据项5所述的方法,其中所述配体包含n-配体基团。
- 还没有人留言评论。精彩留言会获得点赞!
- 噻唑橙苯乙烯类化合物作为G-四链体核酸荧光探针的制作方法与工艺
- 一种氟原子取代的氰基异佛尔酮苯乙烯化合物、晶体化合物及制备方法与流程
- 一种制备反式二苯基乙烯类化合物的方法与流程
- 一种二苯乙烯和二苯乙烷类化合物的磷酰氨基酸及其衍生物的制备与用途的制造方法与工艺
- 制备具有较少丙烯酸的酰胺化合物的手段和方法与流程
- 酸存在下丙烯酸系化合物的SET‑LRP聚合的制造方法与工艺
- 一种二官能烯基苯氧基化合物及其制备方法和利用其改性的可溶性双马来酰亚胺树脂与流程
- 二苯乙烯型化合物、其中间体、制备方法和应用与流程
- 一种基于四苯乙烯的荧光化合物及其制备方法和应用与制造工艺
- 基于二苯乙烯单元的树枝状化合物及有机电致发光器件的制造方法与工艺