基于膦杂芳基衍生物的热激发延迟荧光主体材料及其制备方法和应用与流程
本发明涉及一种热激发延迟荧光主体材料及其制备方法和应用。
背景技术:
有机发光二极管由于其具有启动电压低,发光效率高等优点,因而引起了人们的广泛的研究,作为第三代平面显示和照明技术,有机电致发光二极管目前普遍采用磷光染料来构建磷光电致发光,但由于磷光染料的构建常伴有贵金属存在,这就提高了制备成本,同时对环境也不够友好。因此,人们迫切需要其它类型的染料替代磷光染料。而近期,被称为第三代的有机电致发光技术的热激发延迟荧光技术,已经取得了很大的研究进展,热激发延迟荧光染料可以通过自身三线态到单线态反向隙间窜跃使三线态激子转化为单线态激子,进而利用其发光,理论上可以实现100%的内量子效率。用该类染料制备的器件,同样具有较高的效率,可以和磷光器件相媲美,同时热激发延迟荧光染料均为纯有机化合物,制备成本低,对环境友好。热激发延迟荧光器件的主体材料,为了实现高的器件效率,需要具备以下三点:1、具有较高的三线态能级,使能量很好的从主体传递给客体;2、良好的载流子传输能力,以提高器件电子的注入与传输性能;3、具有较弱的分子间作用力,防止主体分子间主客体分子间的强分子间相互作用引起的发射猝灭。通常,平面类热激发延迟荧光染料分子由于其分子间具有较强的相互作用可以有效地促进载流子传输能力,然而,由于其较强的分子间作用力也会促进分子的聚集,这就会使发射猝灭,降低了器件效率。
技术实现要素:
本发明是要解决现有的平面类热激发延迟荧光染料分子易发生猝灭,导致器件效率低的技术问题,而提供基于膦杂芳基衍生物的热激发延迟荧光主体材料及其制备方法和应用。
本发明的基于膦杂芳基衍生物的热激发延迟荧光主体材料的结构式为其中R=Cz或t-Cz;
当R=Cz时记为4CzDPDPA;
当R=t-Cz时记为4tCzDPDPA;
或者结构式为其中R=Cz或t-Cz;
当R=Cz时记为4CzDPDPSA;
当R=t-Cz时记为4tCzDPDPSA;
或者结构式为其中R=Cz或t-Cz;X=S或O;Y=S或O;
当R=Cz、X=Y=S时记为4CzDPDPS2A;当R=t-Cz、X=Y=S时记为4tCzDPDPS2A;
当R=Cz、X=S、Y=O时记为4CzDPDPSOA;当R=t-Cz、X=S、Y=O时记为4tCzDPDPSOA;
当R=Cz、X=Y=O时记为4CzDPDPO2A;当R=t-Cz、X=Y=O时记为4tCzDPDPO2A;
上述的Cz为基团t-Cz为基团
上述的基于膦杂芳基衍生物的热激发延迟荧光主体材料的制备方法如下:
将邻二溴苯的衍生物、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例加入到四氢呋喃中,在氩气保护下,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体干燥后,直接纯化或者硫化/和氧化后再纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料;其中邻二溴苯的衍生物为1,2-二溴-4,5-二咔唑苯或1,2-二溴-4,5-二叔丁基咔唑苯。
本发明的基于膦杂芳基衍生物的热激发延迟荧光主体材料的结构式还可以为:
其中R=Cz或t-Cz;R’=H或POPh2;
当R=Cz、R’=H时记为4CzPPDPO;当R=t-Cz、R’=H时记为4tCzPPDPO;
当R=Cz、R’=POPh2时记为4CzPPTPO当R=t-Cz、R’=POPh2时4tCzPPTPO;
其中Cz为基团t-Cz为基团POPh2为基团
上述的基于膦杂芳基衍生物的热激发延迟荧光主体材料的制备方法如下:
将邻二溴苯的衍生物、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例加入到四氢呋喃中,在氩气保护下,在温度为-60~-85℃的条件下反应1~3小时;再加入二苯基氯化膦,在温度为-60~-85℃的条件下反应1~3小时;其中二苯基氯化膦与邻二溴苯的衍生物的摩尔比为(1~1.5):1;反应完成后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体干燥后,再经氧化、纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料;其中邻二溴苯的衍生物为1,2-二溴-4,5-二咔唑苯或1,2-二溴-4,5-二叔丁基咔唑苯。
上述的基于膦杂芳基衍生物的热激发延迟荧光主体材料的应用,是将其用于热激发延迟荧光电致发光器件中。
利用基于膦杂芳基衍生物的热激发延迟荧光主体材料制备热激发延迟荧光(TADF)电致发光器件的方法,按以下步骤实现:
一、将经过去离子水清洗的玻璃或塑料衬底放入真空蒸镀仪,真空度为1×10-6mbar,蒸镀速率设为0.1nm s-1,在玻璃或塑料衬底上蒸镀材料为氧化铟锡、厚度为100nm的阳极导电层;
二、在阳极导电层上蒸镀材料为MoO3、厚度为5~30nm的空穴注入层;
三、在空穴注入层上蒸镀材料为N,N′-二苯基-N,N′-(1-萘基)-1,1′-联苯-4,4′-二胺(NPB)、厚度为40~70nm nm的空穴传输层;
四、在空穴传输层上蒸镀材料为1,3-二-9-咔唑基苯(mCP)、厚度为5~20nm的电子阻挡层;
五、在电子阻挡层上继续蒸镀厚度为20~40nm的主体材料和客体材料作为发光层,其中发光层中客体材料掺杂的质量浓度为5%~15%;其中主体材料为基于膦杂芳基衍生物的热激发延迟荧光主体材料;客体材料为2,3,5,6-四(3,6-二苯基-9-咔唑基)-对苯二腈(4CzTPNPh);
六、在发光层上蒸镀材料为二[2-((氧代)二苯基膦基)苯基]醚(DPEPO)、厚度为5~20nm的空穴阻挡层;
七、在空穴阻挡层上蒸镀材料为4,7-二苯基-1,10-菲罗啉(Bphen)、厚度为20~50nm的电子传输层;
八、在电子传输层上蒸镀材料为氟化锂(LiF)、厚度为1~30nm的电子注入层;
九、在电子注入层上蒸镀材料为金属铝(Al)、厚度为100~200nm的阴极导电层,封装,得到热激发延迟荧光(TADF)电致发光器件。
本发明构建了用于热激发延迟荧光器件的基于膦杂芳基衍生物的主体材料,该材料是在芳基上引入磷原子以及进一步硫化或/和氧化得到的稳定的主体材料。选择膦杂芳基衍生物作为主体材料,首先,作为含有膦原子-杂原子分子的材料,该主体分子具有较高的三线态能级,同时该类主体具有平面结构,P=O或P=S基团具有极化分子的作用,提高了分子的电子注入传输能力,使得该类分子材料具有良好的载流子传输能力,另一方面,该类分子通过其上的磷原子、及进一步修饰(氧化及硫化)调节了分子的几何构型,进而达到抑制分子间的相互作用,抑制猝灭,使主体材料在具有良好的载流子传输能力同时还能有效抑制分子间的聚集。从而有效地提高了热激发延迟荧光器件的效率。总之,本发明中膦杂芳基衍生物主体材料用于电致发光器件包含以下优点:
1、保持较高的三线态能级,保证能量从主体到客体的有效传递。
2、提高电致发光器件材料的载流子注入和传输能力,基于膦杂芳基衍生物主体材料制备的电致发光器件将启亮电压降低到3.5~4.2V,具有良好的热力学稳定性,裂解温度为312℃-454℃,同时提高了有机电致发光材料的发光效率和亮度,本发明主要应用于有机电致发光二极管器件中。
附图说明
图1是实施例一合成的4CzDPDPA紫外荧光光谱谱图,其中用■曲线表示4CzDPDPA/二氯甲烷的紫外吸收光谱图,用□曲线表示4CzDPDPA/二氯甲烷的荧光发射光谱图;
图2是实施例一合成的4CzDPDPA的热重分析谱图;
图3是实施例二合成的化合物4CzDPDPSA紫外荧光光谱谱图,其中用■曲线表示4CzDPDPSA/二氯甲烷的紫外吸收光谱图,用□曲线表示4CzDPDPSA/二氯甲烷的荧光发射光谱图;
图4是实施例二合成的4CzDPDPSA的热重分析谱图;
图5是实施例三合成的化合物4CzDPDPS2A紫外荧光光谱谱图,其中用■曲线表示4CzDPDPS2A/二氯甲烷的紫外吸收光谱图,用□曲线表示4CzDPDPS2A/二氯甲烷的荧光发射光谱图;
图6是实施例三合成的4CzDPDPS2A的热重分析谱图;
图7是实施例四合成的化合物4CzDPDPSOA紫外荧光光谱谱图,其中用■曲线表示4CzDPDPSOA/二氯甲烷的紫外吸收光谱图,用□曲线表示4CzDPDPSOA/二氯甲烷的荧光发射光谱图;
图8是实施例四合成的4CzDPDPSOA的热重分析谱图;
图9是实施例五合成的化合物4CzDPDPO2A紫外荧光光谱谱图,其中用■曲线表示4CzDPDPO2A/二氯甲烷的紫外吸收光谱图,用□曲线表示4CzDPDPO2A/二氯甲烷的荧光发射光谱图;
图10是实施例五合成的4CzDPDPO2A的热重分析谱图;
图11是应用实施例一的热激发延迟荧光器件的电压-电流密度关系曲线,其中用■表示4CzDPDPA,用●表示4CzDPDPSA,用▲表示4CzDPDPS2A,用▼表示4CzDPDPSOA,用◆表示4CzDPDPO2A;
图12是应用实施例一的热激发延迟荧光器件的电压-亮度关系曲线,其中用■表示4CzDPDPA,用●表示4CzDPDPSA,用▲表示4CzDPDPS2A,用▼表示4CzDPDPSOA,用◆表示4CzDPDPO2A;
图13是应用实施例一的热激发延迟荧光器件的亮度-电流效率关系曲线,其中用■表示4CzDPDPA,用●表示4CzDPDPSA,用▲表示4CzDPDPS2A,用▼表示4CzDPDPSOA,用◆表示4CzDPDPO2A;
图14是应用实施例一的热激发延迟荧光器件的亮度-功率效关系曲线,其中用■表示4CzDPDPA,用●表示4CzDPDPSA,用▲表示4CzDPDPS2A,用▼表示4CzDPDPSOA,用◆表示4CzDPDPO2A;
图15是应用实施例一的热激发延迟荧光器件的亮度-外量子效率关系曲线,其中用■表示4CzDPDPA,用●表示4CzDPDPSA,用▲表示4CzDPDPS2A,用▼表示4CzDPDPSOA,用◆表示4CzDPDPO2A;
图16是应用实施例一的热激发延迟荧光器件的电致发光光谱,其中用■表示4CzDPDPA,用●表示4CzDPDPSA,用▲表示4CzDPDPS2A,用▼表示4CzDPDPSOA,用◆表示4CzDPDPO2A;
图17是实施例六合成的化合物4tCzDPDPA紫外荧光光谱谱图,其中用■曲线表示4tCzDPDPA/二氯甲烷的紫外吸收光谱图,用□曲线表示4tCzDPDPA/二氯甲烷的荧光发射光谱图;
图18是实施例六合成的4tCzDPDPA的热重分析谱图;
图19是实施例七合成的化合物4tCzDPDPSA紫外荧光光谱谱图,其中用■曲线表示4tCzDPDPSA/二氯甲烷的紫外吸收光谱图,用□曲线表示4tCzDPDPSA/二氯甲烷的荧光发射光谱图;
图20是实施例七合成的4tCzDPDPSA的热重分析谱图;
图21是实施例八合成的化合物4tCzDPDPS2A紫外荧光光谱谱图,其中用■曲线表示4tCzDPDPS2A/二氯甲烷的紫外吸收光谱图,用□曲线表示4tCzDPDPS2A/二氯甲烷的荧光发射光谱图;
图22是实施例八合成的4tCzDPDPS2A的热重分析谱图;
图23是实施例九合成的化合物4tCzDPDPSOA紫外荧光光谱谱图,其中用■曲线表示4tCzDPDPSOA/二氯甲烷的紫外吸收光谱图,用□曲线表示4tCzDPDPSOA/二氯甲烷的荧光发射光谱图;
图24是实施例九合成的4tCzDPDPSOA的热重分析谱图;
图25是实施例十合成的化合物4tCzDPDPO2A紫外荧光光谱谱图,其中用■曲线表示4tCzDPDPO2A/二氯甲烷的紫外吸收光谱图,用□曲线表示4tCzDPDPO2A/二氯甲烷的荧光发射光谱图;
图26是实施例十合成的4tCzDPDPO2A的热重分析谱图;
图27是应用实施例二的热激发延迟荧光器件的电压-电流密度关系曲线,其中用□表示4tCzDPDPA,用○表示4tCzDPDPSA,用△表示4tCzDPDPS2A,用▽表示4tCzDPDPSOA,用◇表示4tCzDPDPO2A;
图28是应用实施例二的热激发延迟荧光器件的电压-亮度关系曲线,其中用□表示4tCzDPDPA,用○表示4tCzDPDPSA,用△表示4tCzDPDPS2A,用▽表示4tCzDPDPSOA,用◇表示4tCzDPDPO2A;
图29是应用实施例二的热激发延迟荧光器件的亮度-电流效率关系曲线,其中用□表示4tCzDPDPA,用○表示4tCzDPDPSA,用△表示4tCzDPDPS2A,用▽表示4tCzDPDPSOA,用◇表示4tCzDPDPO2A;
图30是应用实施例二的热激发延迟荧光器件的亮度-功率效率关系曲线,其中用□表示4tCzDPDPA,用○表示4tCzDPDPSA,用△表示4tCzDPDPS2A,用▽表示4tCzDPDPSOA,用◇表示4tCzDPDPO2A;
图31是应用实施例二的热激发延迟荧光器件的亮度-外量子效率关系曲线,其中用□表示4tCzDPDPA,用○表示4tCzDPDPSA,用△表示4tCzDPDPS2A,用▽表示4tCzDPDPSOA,用◇表示4tCzDPDPO2A;
图32是应用实施例二的热激发延迟荧光器件的电致发光光谱,其中用□表示4tCzDPDPA,用○表示4tCzDPDPSA,用△表示4tCzDPDPS2A,用▽表示4tCzDPDPSOA,用◇表示4tCzDPDPO2A。
图33是实施例十一合成的4CzPPDPO紫外荧光光谱谱图,其中用■曲线表示4CzPPDPO/二氯甲烷的紫外吸收光谱图,用□曲线表示4CzPPDPO/二氯甲烷的荧光发射光谱图;
图34是实施例十一合成的4CzPPDPO的热重分析谱图;
图35是实施例十二合成的化合物4CzPPTPO紫外荧光光谱谱图,其中用■曲线表示4Cz DPDPSA/二氯甲烷的紫外吸收光谱图,用□曲线表示4CzPPTPO/二氯甲烷的荧光发射光谱图;
图36是实施例十二合成的4CzPPTPO的热重分析谱图;
图37是实施例十三合成的化合物4tCzPPDPO紫外荧光光谱谱图,其中用■曲线表示DPDPS2A/二氯甲烷的紫外吸收光谱图,用□曲线表示4tCzPPDPO/二氯甲烷的荧光发射光谱图;
图38是实施例十三合成的4tCzPPDPO的热重分析谱图;
图39是实施例十四合成的化合物4tCzPPTPO紫外荧光光谱谱图,其中用■曲线表示4tCzPPTPO/二氯甲烷的紫外吸收光谱图,用□曲线表示4tCzPPTPO/二氯甲烷的荧光发射光谱图;
图40是实施例十四合成的4tCzPPTPO的热重分析谱图;
图41是应用实施例三的热激发延迟荧光器件的电压-电流密度关系曲线,其中用■表示4CzPPDPO,用●表示4CzPPTPO,用▲表示4tCzPPDPO,用▼表示4tCzPPTPO;
图42是应用实施例三的热激发延迟荧光器件的电压-亮度关系曲线,其中用■表示4CzPPDPO,用●表示4CzPPTPO,用▲表示4tCzPPDPO,用▼表示4tCzPPTPO;
图43是应用实施例三的热激发延迟荧光器件的亮度-电流效率关系曲线,其中用■表示4CzPDPA,用●表示4CzPPTPO,用▲表示4tCzPPDPO,用▼表示4tCzPPTPO;
图44是应用实施例三的热激发延迟荧光器件的亮度-功率效关系曲线,其中用■表示4CzPPDPO,用●表示4CzPPTPO,用▲表示4tCzPPDPO,用▼表示4tCzPPTPO;
图45是应用实施例三的热激发延迟荧光器件的亮度-外量子效率关系曲线,其中用■表示4CzPPDPO,用●表示4CzPPTPO,用▲表示4tCzPPDPO,用▼表示4Cz DPDPSOA;
图46是应用实施例三的热激发延迟荧光器件的电致发光光谱,其中用■表示4CzPPDPO,用●表示4CzPPTPO,用▲表示4tCzPPDPO,用▼表示4tCzPPTPO。
具体实施方式
具体实施方式一:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料的结构式为其中R=Cz或t-Cz;
当R=Cz时记为4CzDPDPA;
当R=t-Cz时记为4tCzDPDPA;
或者结构式为其中R=Cz或t-Cz;
当R=Cz时记为4CzDPDPSA;
当R=t-Cz时记为4tCzDPDPSA;
或者结构式为其中R=Cz或t-Cz;X=S或O;Y=S或O;
当R=Cz、X=Y=S时记为4CzDPDPS2A;当R=t-Cz、X=Y=S时记为4tCzDPDPS2A;
当R=Cz、X=S、Y=O时记为4CzDPDPSOA;当R=t-Cz、X=S、Y=O时记为4tCzDPDPSOA;
当R=Cz、X=Y=O时记为4CzDPDPO2A;当R=t-Cz、X=Y=O时记为4tCzDPDPO2A;
上述的Cz为基团t-Cz为基团
具体实施方式二:具体实施方式一所述的基于膦杂芳基衍生物的热激发延迟荧光主体材料的制备方法如下:
将邻二溴苯的衍生物、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例加入到四氢呋喃中,在氩气保护下,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体干燥后,直接纯化或者硫化/和氧化后再纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料;其中邻二溴苯的衍生物为1,2-二溴-4,5-二咔唑苯或1,2-二溴-4,5-二叔丁基咔唑苯。
具体实施方式三:本实施方式与具体实施方式二不同的是所述的硫化过程是:向有机层液体中加入硫粉,在温度为15~30℃的条件下搅拌进行反应0.5~2小时,完成硫化。其它与具体实施方式二相同。
具体实施方式四:本实施方式与具体实施方式二或三不同的是所述的氧化的过程是:向有机层液体中加入H2O2,在温度为-5~20℃的条件下搅拌进行反应0.5~2小时,完成氧化。其它与具体实施方式二或三相同。
具体实施方式五:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPA的合成方法如下:将1,2-二溴-4,5-二咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,旋转蒸发去除二氯甲烷溶剂,以石油醚和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPA;其中1,2-二溴-4,5-二咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml。
具体实施方式六:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPSA的合成方法如下:将1,2-二溴-4,5-二咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,加入硫粉,在温度为15~30℃的条件下搅拌进行硫化反应0.5~2小时,抽滤、旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPSA;其中1,2-二溴-4,5-二咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml;加入的硫粉的物质量的量与苯基二氯化膦的物质的量的比为(0.5~0.75):2。
具体实施方式七:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPS2A的合成方法如下:将1,2-二溴-4,5-二咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,加入硫粉,在温度为15~30℃的条件下搅拌进行硫化反应1~2小时,抽滤、旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPS2A;其中1,2-二溴-4,5-二咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml;加入的硫粉的物质量的量与苯基二氯化膦的物质的量的比为(0.5~0.75):1。
具体实施方式八:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPSOA的合成方法如下:将1,2-二溴-4,5-二咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,加入硫粉,在温度为15~30℃的条件下搅拌进行硫化反应1~2小时;再加入H2O2,在温度为-5~20℃的条件下搅拌进行氧化反应0.5~2小时;用亚硫酸氢钠溶液和水分别洗涤,二氯甲烷萃取,无水硫酸钠干燥后,旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPSOA;其中1,2-二溴-4,5-二咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml;加入的硫粉的物质量的量与苯基二氯化膦的物质的量的比为(0.5~0.75):2;加入的H2O2与苯基二氯化膦的物质的量的比(10~15):1。
具体实施方式九:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPO2A的合成方法如下:将1,2-二溴-4,5-二咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,加入H2O2,在温度为-5~20℃的条件下搅拌进行氧化反应0.5~2小时;用亚硫酸氢钠溶液和水分别洗涤,二氯甲烷萃取,无水硫酸钠干燥后,旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPO2A;其中1,2-二溴-4,5-二咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml;加入的H2O2与苯基二氯化膦的物质的量的比(20~25):1。
具体实施方式十:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPA的合成方法如下:
将1,2-二溴-4,5-二叔丁基咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,旋转蒸发去除二氯甲烷溶剂,以石油醚和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPA;其中1,2-二溴-4,5-二叔丁基咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml。
具体实施方式十一:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPSA的合成方法如下:
将1,2-二溴-4,5-二叔丁基咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,加入硫粉,在温度为15~30℃的条件下搅拌进行硫化反应0.5~2小时,抽滤、旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPSA;其中1,2-二溴-4,5-二叔丁基咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml;加入的硫粉的物质量的量与苯基二氯化膦的物质的量的比为(0.5~0.75):2
具体实施方式十二:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPS2A的合成方法如下:
将1,2-二溴-4,5-二叔丁基咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,加入硫粉,在温度为15~30℃的条件下搅拌进行硫化反应1~2小时,抽滤、旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPS2A;其中1,2-二溴-4,5-二叔丁基咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml;加入的硫粉的物质量的量与苯基二氯化膦的物质的量的比为(0.5~0.75):1。
具体实施方式十三:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPSOA的合成方法如下:
将1,2-二溴-4,5-二叔丁基咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,加入硫粉,在温度为15~30℃的条件下搅拌进行硫化反应1~2小时;再加入H2O2,在温度为-5~20℃的条件下搅拌进行氧化反应0.5~2小时;用亚硫酸氢钠溶液和水分别洗涤、用二氯甲烷萃取、用无水硫酸钠干燥后,旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPSOA;其中1,2-二溴-4,5-二叔丁基咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml;加入的硫粉的物质量的量与苯基二氯化膦的物质的量的比为(0.5~0.75):2;加入的H2O2与苯基二氯化膦的物质的量的比(10~15):1;
具体实施方式十四:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPO2A的合成方法如下:将1,2-二溴-4,5-二叔丁基咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,加入H2O2,在温度为-5~20℃的条件下搅拌进行氧化反应0.5~2小时;用亚硫酸氢钠溶液和水分别洗涤,二氯甲烷萃取,无水硫酸钠干燥后,旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPO2A;其中1,2-二溴-4,5-二叔丁基咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml;加入的H2O2与苯基二氯化膦的物质的量的比(20~25):1。
具体实施方式十五:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料的结构式为:
其中R=Cz或t-Cz;R’=H或POPh2;
当R=Cz、R’=H时记为4CzPPDPO;当R=t-Cz、R’=H时记为4tCzPPDPO;
当R=Cz、R’=POPh2时记为4CzPPTPO当R=t-Cz、R’=POPh2时4tCzPPTPO;
其中Cz为基团t-Cz为基团POPh2为基团
具体实施方式十六:具体实施方式十五所述的基于膦杂芳基衍生物的热激发延迟荧光主体材料的制备方法如下:
将邻二溴苯的衍生物、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例加入到四氢呋喃中,在氩气保护下,在温度为-60~-85℃的条件下反应1~3小时;再加入二苯基氯化膦,在温度为-60~-85℃的条件下反应1~3小时;其中二苯基氯化膦与邻二溴苯的衍生物的摩尔比为(1~1.5):1;反应完成后倒入水中,用二氯甲烷萃取,得到有机层,再经氧化、纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料;其中邻二溴苯的衍生物为1,2-二溴-4,5-二咔唑苯或1,2-二溴-4,5-二叔丁基咔唑苯。
具体实施方式十七:本实施方式与具体实施方式十六不同的是氧化的过程是:向有机层液体中加入H2O2,在温度为-5~20℃的条件下搅拌进行反应0.5~2小时,完成氧化。其它与具体实施方式十六相同。
具体实施方式十八:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzPPDPO的制备方法如下:
基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzPPDPO的合成方法如下:将1,2-二溴-4,5-二咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时,按照二苯基氯化膦与1,2-二溴-4,5-二咔唑苯的摩尔比为(1~1.5):1加入二苯基氯化膦,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,向有机层中加入H2O2,在温度为-5~20℃的条件下搅拌进行氧化反应0.5~2小时;用亚硫酸氢钠溶液和水分别洗涤,二氯甲烷萃取,无水硫酸钠干燥后,旋转蒸发除去二氯甲烷溶剂,以乙醇和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzPPDPO;其中1,2-二溴-4,5-二咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml。
具体实施方式十八:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzPPTPO的制备方法如下:
基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzPPTPO的合成方法如下:将1,2-二溴-4,5-二咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时;再按照二苯基氯化膦与1,2-二溴-4,5-二咔唑苯的摩尔比为(1.6~3):1加入二苯基氯化膦,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,向有机层中加入H2O2,在温度为-5~20℃的条件下搅拌进行氧化反应0.5~2小时;用亚硫酸氢钠溶液和水分别洗涤,再用二氯甲烷萃取,无水硫酸钠干燥后,,旋转蒸发除去二氯甲烷溶剂,再以乙醇和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzPPTPO;其中1,2-二溴-4,5-二咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml。
具体实施方式十九:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzPPDPO的制备方法如下:
基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzPPDPO的合成方法如下:将1,2-二溴-4,5-二叔丁基咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时,按照二苯基氯化膦与1,2-二溴-4,5-二咔唑叔丁基苯的摩尔比为(1~1.5):1加入二苯基氯化膦,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,向有机层中加入H2O2,在温度为-5~20℃的条件下搅拌进行氧化反应0.5~2小时;用亚硫酸氢钠溶液和水分别洗涤,再用二氯甲烷萃取,无水硫酸钠干燥后,旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzPPDPO;其中1,2-二溴-4,5-二叔丁基咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml;
具体实施方式二十:本实施方式的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzPPTPO的制备方法如下:
基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzPPTPO的合成方法如下:将1,2-二溴-4,5-二叔丁基咔唑苯、苯基二氯化膦、正丁基锂按摩尔比为1:(1~1.5):(2~4)的比例混于四氢呋喃加入反应器中,在温度为-60~-85℃的条件下反应1~3小时;再按照二苯基氯化膦与1,2-二溴-4,5-二叔丁基咔唑苯的摩尔比为(1.6~3):1加入二苯基氯化膦,在温度为-60~-85℃的条件下反应1~3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,向有机层中加入H2O2,在温度为-5~20℃的条件下搅拌进行氧化反应0.5~2小时;用亚硫酸氢钠溶液和水分别洗涤,二氯甲烷萃取,无水硫酸钠干燥后,旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzPPTPO;其中1,2-二溴-4,5-二叔丁基咔唑苯的物质的量与四氢呋喃的体积的比为1mmol:(10~25)ml。
具体实施方式二十一:具体实施方式一所述的基于膦杂芳基衍生物的热激发延迟荧光主体材料的应用,是将其用于热激发延迟荧光电致发光器件中。
具体实施方式二十二:利用基于膦杂芳基衍生物的热激发延迟荧光主体材料制备热激发延迟荧光(TADF)电致发光器件的方法,按以下步骤实现:
一、将经过去离子水清洗的玻璃或塑料衬底放入真空蒸镀仪,真空度为1×10-6mbar,蒸镀速率设为0.1nm s-1,在玻璃或塑料衬底上蒸镀材料为氧化铟锡、厚度为100nm的阳极导电层;
二、在阳极导电层上蒸镀材料为MoO3、厚度为5~30nm的空穴注入层;
三、在空穴注入层上蒸镀材料为N,N′-二苯基-N,N′-(1-萘基)-1,1′-联苯-4,4′-二胺(NPB)、厚度为40~70nm nm的空穴传输层;
四、在空穴传输层上蒸镀材料为1,3-二-9-咔唑基苯(mCP)、厚度为5~20nm的电子阻挡层;
五、在电子阻挡层上继续蒸镀厚度为20~40nm的主体材料和客体材料作为发光层,其中发光层中客体材料掺杂的质量浓度为5%~15%;其中主体材料为基于膦杂芳基衍生物的热激发延迟荧光主体材料;客体材料为2,3,5,6-四(3,6-二苯基-9-咔唑基)-对苯二腈(4CzTPNPh);
六、在发光层上蒸镀材料为二[2-((氧代)二苯基膦基)苯基]醚(DPEPO)、厚度为5~20nm的空穴阻挡层;
七、在空穴阻挡层上蒸镀材料为4,7-二苯基-1,10-菲罗啉(Bphen)、厚度为20~50nm的电子传输层;
八、在电子传输层上蒸镀材料为氟化锂(LiF)、厚度为1~30nm的电子注入层;
九、在电子注入层上蒸镀材料为金属铝(Al)、厚度为100~200nm的阴极导电层,封装,得到热激发延迟荧光(TADF)电致发光器件。
用以下实施例验证本发明的有益效果:
实施例一:本实施例的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPA的合成方法按下列步骤进行:
一、将1mmol 1,2-二溴-4,5-二咔唑苯、1mmol的苯基二氯化膦、2mmol正丁基锂加入到盛有20ml的THF的50ml三颈圆底烧瓶中,在氩气保护下,在温度为-78℃条件下搅拌2小时后倒入水中,再用二氯甲烷萃取,得到有机层,有机层液体用无水NaSO4干燥后,减压蒸馏除去二氯甲烷溶剂,再以石油醚与二氯甲烷的体积比6:1的混合溶剂为淋洗剂进行柱层析纯化,得到4CzDPDPA。
本实施例制备的4CzDPDPA,质谱仪测得数据为:m/z:1028.32(100.0%),1029.32(79.4%),1030.33(30.3%),1031.33(7.7%),1032.33(1.5%),1030.32(1.2%);GC-MS:m/z(%):1028(100)[M+];Elemental Analysis of C72H46N4P2:C,84.03;H,4.51;N,5.44。从而可知4CzDPDPA的结构式为:
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPA的紫外荧光光谱谱图如图1所示。
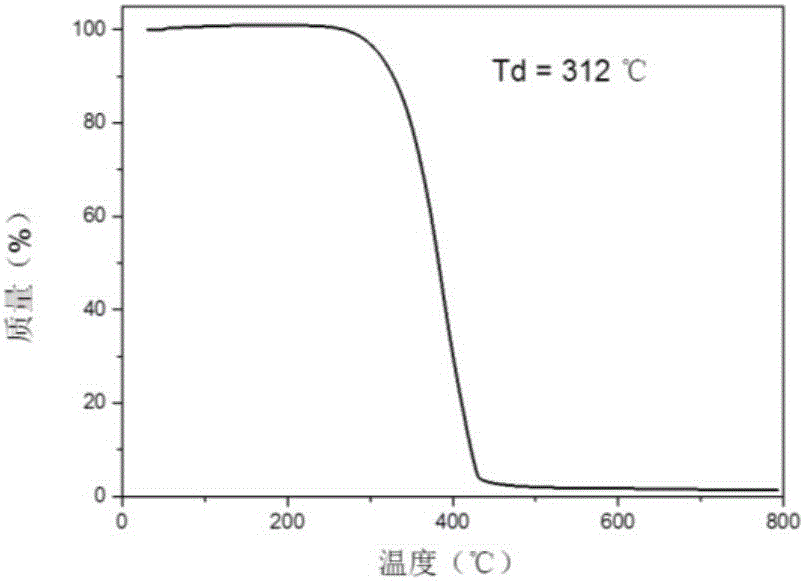
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPA的热重分析谱图如图2所示,由图2可知4CzDPDPA的裂解温度达312℃。
实施例二:本实施例的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPSA的合成方法按下列步骤进行:
一、将1mmol1,2-二溴-4,5-二咔唑苯、1.2mmol的苯基二氯化膦、2mmol正丁基锂加入到装有20ml的THF的50ml的三颈圆底烧瓶中,在氩气保护下,在温度为-78℃下搅拌2小时后倒入水中,再用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,向有机层液体中加入0.4mmol硫粉,在20℃条件下搅拌0.5小时进行硫化,抽滤、旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷体积比1:20的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPSA。
本实施例制备的4CzDPDPSA,质谱仪测得数据为:m/z:1060.29(100.0%),1061.30(78.4%),1062.30(30.3%),1063.30(8.4%),1062.29(6.3%),1063.29(3.6%),1061.29(2.3%),1064.31(1.4%),1064.29(1.4%);GC-MS:m/z(%):1061(100)[M+];Elemental Analysis of C72H46N4P2S:C,81.49;H,4.37;N,5.28;S,3.02。从而可知,4CzDPDPSA的结构式为
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPSA的紫外荧光光谱谱图如图3所示。
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPSA的热重分析谱图如图4所示,由图4可知4CzDPDPSA的裂解温度达362℃。
实施例三:本实施例的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPS2A的合成方法按下列步骤进行:
一、将1mmol 1,2-二溴-4,5-二咔唑苯、1.2mmol的苯基二氯化膦、2mmol正丁基锂和20ml的THF混合于50ml三颈圆底烧瓶中,在氩气保护下,在温度为-78℃下搅拌3小时后倒入水中,再用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,加入0.6mmol硫粉,在温度为20℃的条件下搅拌进行硫化反应1小时,抽滤、旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷体积比1:20的混合溶剂为淋洗剂柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPS2A。
本实施例制备的4tCzPPDPO,质谱仪测得数据为:m/z:1092.26(100.0%),1093.27(78.4%),1094.27(31.6%),1094.26(10.2%),1095.27(8.5%),1095.26(7.3%),1093.26(3.1%),1096.27(3.0%),1096.28(1.4%);GC-MS:m/z(%):1093(100)[M+];Elemental Analysis of C72H46N4P2S2:C,79.10;H,4.24;N,5.12;S,5.87。从而可知,4CzDPDPS2A的结构式为
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPS2A的紫外荧光光谱谱图如图5所示。
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPS2A的热重分析谱图如图6所示,由图6可知4CzDPDPS2A的裂解温度达367℃。
实施例四:本实施例基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPSOA的合成方法按下列步骤进行:
将1mmol 1,2-二溴-4,5-二咔唑苯、1.2mmol的苯基二氯化膦、2mmol正丁基锂和20ml的THF混合于50ml三颈圆底烧瓶中,在氩气保护下,在温度为-78℃下搅拌1~3小时后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,加入0.35mmol硫粉,在温度为20℃的条件下搅拌进行硫化反应1小时;再加入12mmolH2O2,在温度为0℃的条件下搅拌进行氧化反应1小时;用亚硫酸氢钠溶液和水分别洗涤,二氯甲烷萃取,无水硫酸钠干燥后,旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷体积比1:20的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPSOA;
本实施例制备的4CzDPDPSOA,质谱仪测得数据为:m/z:1076.29(100.0%),1077.29(79.2%),1078.29(31.9%),1079.30(7.7%),1078.28(4.5%),1079.29(4.4%),1080.30(1.6%),1080.29(1.5%),1077.28(1.5%);GC-MS:m/z(%):1077(100)[M+];Elemental Analysis of C72H46N4OP2S:C,80.28;H,4.30;N,5.20;O,1.49;S,2.98。从而可知,本实施例制备的4CzDPDPSOA的结构式为:
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPSOA的紫外荧光光谱谱图如图7所示。
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPSOA的热重分析谱图如图8所示,由图8可知本实施例的4CzDPDPSOA的裂解温度达377℃。
实施例五:本实施例的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPO2A的合成方法按下列步骤进行:
一、将1mmol 1,2-二溴-4,5-二咔唑苯、1.2mmol的苯基二氯化膦、2mmol正丁基锂和20ml的THF混合于50ml三颈圆底烧瓶中,在氩气保护下,在温度为-78℃下搅拌3小时后倒入水中,再用二氯甲烷萃取,得到有机层,加入25mmol的H2O2,在温度为0℃的条件下搅拌进行氧化反应1小时;用亚硫酸氢钠溶液和水分别洗涤,二氯甲烷萃取,无水硫酸钠干燥后,旋转蒸发去除二氯甲烷溶剂,以乙醇和二氯甲烷体积比1:20的混合溶剂为淋洗剂柱层析纯化,得到4CzDPDPO2A。
本实施例制备的4CzDPDPO2A,质谱仪测得数据为:m/z:1060.31(100.0%),1061.31(79.4%),1062.32(30.4%),1063.32(8.1%),1064.32(1.7%),1062.31(1.6%);GC-MS:m/z(%):1061(100)[M+];Elemental Analysis of C72H46N4O2P2:C,81.50;H,4.37;N,5.28;O,3.02。从而可知4CzDPDPO2A的结构式为:
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPO2A的紫外荧光光谱谱图如图9所示。
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPO2A的热重分析谱图如图10所示,由图10可知4CzDPDPO2A的裂解温度达405℃。
应用实施例一:将实施例一至五制备的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzDPDPA、4CzDPDPSA、4CzDPDPS2A、4CzDPDPSOA、4CzDPDPO2A分别用于制备电致发光器件,具体按以下步骤进行:
一、将经过去离子水清洗的玻璃或塑料衬底放入真空蒸镀仪,真空度为1×10-6mbar,蒸镀速率设为0.1nm s-1,在玻璃或塑料衬底上蒸镀材料为氧化铟锡,厚度为100nm的阳极导电层;
二、在阳极导电层上蒸镀材料为MoO3,厚度为20nm的空穴注入层;
三、在空穴注入层上蒸镀材料为NPB,厚度为50nm的空穴传输层;
四、在空穴传输层上蒸镀材料为mCP,厚度为10nm的电子阻挡层;
五、在电子阻挡层上继续蒸镀厚度为30nm的主体材料(基于膦杂芳基衍生物的热激发延迟荧光主体材料)和客体材料客体材料为2,3,5,6-四(3,6-二苯基-9-咔唑基)-对苯二腈(4CzTPNPh);
六、在发光层上蒸镀材料为DPEPO,厚度为15nm的空穴阻挡层;
七、在空穴阻挡层上蒸镀材料为Bphen,厚度为40nm的电子传输层;
八、在电子传输层上蒸镀材料为LiF,厚度为20nm的电子注入层;
九、在电子注入层上蒸镀材料为金属Al,厚度为150nm的阴极导电层,封装,得到热激发延迟荧光(TADF)电致发光器件。
本应用实施例制备的电致发光器件的电压-电流密度关系曲线如图11所示。其中的■表示4CzDPDPA,●表示4CzDPDPSA,▲表示4CzDPDPS2A,▼表示4CzDPDPSOA,◆表示4CzDPDPO2A。从图11可以看出,随着电压的升高,器件的电流密度都呈现出升高的趋势。
本应用实施例的电致发光器件的电压-亮度关系曲线如图12所示,其中的■表示4CzDPDPA,●表示4CzDPDPSA,▲表示4CzDPDPS2A,▼表示4CzDPDPSOA,◆表示4CzDPDPO2A。由此图可知以4CzDPDPA制备的电致发光器件的启亮电压为3.6V,以4CzDPDPSA制备的电致发光器件的启亮电压为3.6V,以4CzDPDPS2A制备的电致发光器件的启亮电压为3.6V,以4CzDPDPSOA制备的电致发光器件的启亮电压为3.7V,以4CzDPDPO2A制备的电致发光器件的启亮电压为3.7V。
本应用实施例的电致发光器件的亮度-电流效率关系曲线如图13所示,其中的■表示4CzDPDPA,●表示4CzDPDPSA,▲表示4CzDPDPS2A,▼表示4CzDPDPSOA,◆表示4CzDPDPO2A。从图中可以看出,由此图可知以4CzDPDPA制备的电致发光器件的电流效率达到最大值为17.2cd·A-1,以4CzDPDPSA制备的电致发光器件的电流效率达到最大值为11.6cd·A-1,以4CzDPDPS2A制备的电致发光器件的电流效率达到最大值为7.1cd·A-1,以4CzDPDPSOA制备的电致发光器件的电流效率达到最大值为6.5cd·A-1,以4CzDPDPO2A制备的电致发光器件的电流效率达到最大值为7.7cd·A-1。
本应用实施例的电致发光器件的亮度-功率效率关系曲线如图14所示,其中的■表示4CzDPDPA,●表示4CzDPDPSA,▲表示4CzDPDPS2A,▼表示4CzDPDPSOA,◆表示4Cz DPDPO2A。由此图可知以4Cz DPDPA制备的电致发光器件的功率效率达到最大值为10lm·W-1,以4Cz DPDPSA制备的电致发光器件的功率效率达到最大值为10.4lm·W-1,以4CzDPDPS2A制备的电致发光器件的功率效率达到最大值为3.9lm·W-1,以4CzDPDPSOA制备的电致发光器件的功率效率达到最大值为3.6lm·W-1,以4CzDPDPO2A制备的电致发光器件的功率效率达到最大值为4.2lm·W-1。
本应用实施例的电致发光器件的亮度-外量子效率关系曲线如图15所示,其中■表示4CzDPDPA,●表示4CzDPDPSA,▲表示4CzDPDPS2A,▼表示4CzDPDPSOA,◆表示4CzDPDPO2A,由此图可知以4CzDPDPA制备的电致发光器件的最大外量子效率6.1%,以4CzDPDPSA制备的电致发光器件的最大外量子效率6.3%,以4CzDPDPS2A制备的电致发光器件的最大外量子效率3.9%,以4CzDPDPSOA制备的电致发光器件的最大外量子效率3.6%,以4CzDPDPO2A制备的电致发光器件的的最大外量子效率4.2%。
本应用实施例的电致发光器件的电致发光光谱图如图16所示,■表示4CzDPDPA,●表示4CzDPDPSA,▲表示4CzDPDPS2A,▼表示4CzDPDPSOA,◆表示4CzDPDPO2A,由此图可知以4CzDPDPA制备的电致发光器件的电致发光峰在602nm处、以4CzDPDPSA制备的电致发光器件的电致发光峰在599nm处、以4CzDPDPS2A制备的电致发光器件的电致发光峰在596nm处、以4CzDPDPSOA制备的电致发光器件的电致发光峰在603nm处,以4CzDPDPO2A制备的电致发光器件的电致发光峰在601nm处。
实施例六:本实施例的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPA的合成方法按下列步骤进行:
一、将1mmol 1,2-二溴-4,5-二叔丁基咔唑苯、1mmol的苯基二氯化膦、2mmol正丁基锂加入到盛有20ml的THF的50ml三颈圆底烧瓶中,在氩气保护下,在温度为-78℃条件下搅拌2小时后倒入水中,再用二氯甲烷萃取,得到有机层,有机层液体用无水NaSO4干燥后,旋转蒸发除去二氯甲烷溶剂,再以石油醚与二氯甲烷的体积比6:1的混合溶剂为淋洗剂进行柱层析纯化,得到4tCzDPDPA。
本实施例制备的4tCzDPDPA,质谱仪测得数据为:
m/z:1477.82(100.0%),1476.82(87.7%),1478.83(56.2%),1479.83(20.9%),1480.83(5.8%),1478.82(1.5%),1481.84(1.2%),1477.83(1.1%);GC-MS:m/z(%):1478(100)[M+];Elemental Analysis of C104H110N4P2:C,84.52;H,7.50;N,3.79。从而可知4tCzDPDPA的结构式为:
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPA的紫外荧光光谱谱图如图17所示。
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPA的热重分析谱图如图18所示,由图18可知4tCzDPDPA的裂解温度达317℃。
实施例七:本实施例的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPSA的合成方法按下列步骤进行:
二、将1mmol 1,2-二溴-4,5-二叔丁基咔唑苯、1.2mmol的苯基二氯化膦、2mmol正丁基锂加入到装有20ml的THF的50ml的三颈圆底烧瓶中,在氩气保护下,在温度为-78℃下搅拌2小时后倒入水中,再用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,向有机层液体中加入0.3mmol硫粉,在20℃条件下搅拌0.5小时进行硫化,抽滤、旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷体积比1:20的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPSA。
本实施例制备的4tCzDPDPSA,质谱仪测得数据为:m/z:1509.80(100.0%),1508.79(87.9%),1510.80(57.1%),1511.80(21.5%),1512.81(5.8%),1510.79(5.5%),1511.79(4.6%),1512.80(3.0%),1509.79(2.0%),1513.81(1.3%),1513.80(1.0%);GC-MS:m/z(%):1510(100)[M+];Elemental Analysis of C104H110N4P2S:C,82.72;H,7.34;N,3.71;S,2.12。从而可知,4tCzDPDPSA的结构式为
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPSA的紫外荧光光谱谱图如图19所示。
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPSA的热重分析谱图如图20所示,由图20可知4tCzDPDPSA的裂解温度达368℃。
实施例八:本实施例的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPS2A的合成方法按下列步骤进行:
二、将1mmol 1,2-二溴-4,5-二叔丁基咔唑苯、1.2mmol的苯基二氯化膦、2mmol正丁基锂和20ml的THF混合于50ml三颈圆底烧瓶中,在氩气保护下,在温度为-78℃下搅拌3小时后倒入水中,再用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,加入0.6mmol硫粉,在温度为20℃的条件下搅拌进行硫化反应1小时,抽滤、旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷体积比1:20的混合溶剂为淋洗剂柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPS2A。
本实施例制备的4tCzDPDPS2A,质谱仪测得数据为:m/z:1541.77(100.0%),1540.76(87.9%),1542.77(59.4%),1543.77(22.1%),1543.76(9.2%),1542.76(8.0%),1544.78(5.8%),1544.77(5.7%),1541.76(2.7%),1545.77(1.9%),1545.78(1.4%);GC-MS:m/z(%):1542(100)[M+];Elemental Analysis of C104H110N4P2S2:C,81.00;H,7.19;N,3.63;S,4.16。从而可知,4tCzDPDPS2A的结构式为
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPS2A的紫外荧光光谱谱图如图21所示。
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPS2A的热重分析谱图如图22所示,由图22可知4tCzDPDPS2A的裂解温度达380℃。
实施例九:本实施例基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPSOA的合成方法按下列步骤进行:
将1mmol1,2-二溴-4,5-二叔丁基咔唑苯、1.2mmol的苯基二氯化膦、2mmol正丁基锂和20ml的THF混合于50ml三颈圆底烧瓶中,在氩气保护下,在温度为-78℃下搅拌2小时后倒入水中,用二氯甲烷萃取,得到有机层,把有机层液体用无水硫酸钠干燥后,加入0.35mmol硫粉,在温度为20℃的条件下搅拌进行硫化反应1小时;再加入12mmolH2O2,在温度为0℃的条件下搅拌进行氧化反应1小时;用亚硫酸氢钠溶液和水分别洗涤,二氯甲烷萃取,无水硫酸钠干燥后,再旋转蒸发去除二氯甲烷溶剂,再以乙醇和二氯甲烷体积比1:20的混合溶剂为淋洗剂进行柱层析纯化,得到基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPSOA;
本实施例制备的4tCzDPDPSOA,质谱仪测得数据为:m/z:1525.79(100.0%),1524.79(87.3%),1526.79(57.1%),1527.80(21.0%),1528.80(6.0%),1527.79(5.8%),1526.78(4.0%),1528.79(2.9%),1529.80(1.4%),1525.78(1.3%),1526.80(1.3%);GC-MS:m/z(%):1526(100)[M+];Elemental Analysis of C104H110N4OP2S:C,81.85;H,7.27;N,3.67;O,1.05;S,2.10。从而可知,本实施例制备的4tCzDPDPSOA的结构式为:
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPSOA的紫外荧光光谱谱图如图23所示。
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPSOA的热重分析谱图如图24所示,由图8可知本实施例的4tCzDPDPSOA的裂解温度达391℃。
实施例十:本实施例的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPO2A的合成方法按下列步骤进行:
二、将1mmol 1,2-二溴-4,5-二叔丁基咔唑苯、1.2mmol的苯基二氯化膦、2mmol正丁基锂和20ml的THF混合于50ml三颈圆底烧瓶中,在氩气保护下,在温度为-78℃下搅拌3小时后倒入水中,再用二氯甲烷萃取,得到有机层,加入25mmol的H2O2,在温度为0℃的条件下搅拌进行氧化反应1小时;用亚硫酸氢钠溶液和水分别洗涤,二氯甲烷萃取,无水硫酸钠干燥后,旋转蒸发去除二氯甲烷溶剂,以乙醇和二氯甲烷体积比1:20的混合溶剂为淋洗剂柱层析纯化,得到4tCzDPDPO2A。
本实施例制备的4tCzDPDPO2A,质谱仪测得数据为:m/z:1509.81(100.0%),1508.81(87.7%),1510.82(56.3%),1511.82(21.4%),1512.82(6.1%),1510.81(1.8%),1513.83(1.3%),1509.82(1.1%);GC-MS:m/z(%):1509(100)[M+];Elemental Analysis of C104H110N4O2P2:C,82.72;H,7.34;N,3.71;O,2.12。从而可知4tCzDPDPO2A的结构式为:
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPO2A的紫外荧光光谱谱图如图25所示。
本实施例得到的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPO2A的热重分析谱图如图26所示,由图26可知4tCzDPDPO2A的裂解温度达424℃。
应用实施例二:将实施例一至五制备的基于膦杂芳基衍生物的热激发延迟荧光主体材料4tCzDPDPA、4tCzDPDPSA、4tCzDPDPS2A、4tCzDPDPSOA、4tCzDPDPO2A分别用于制备电致发光器件,具体按以下步骤进行:
一、将经过去离子水清洗的玻璃或塑料衬底放入真空蒸镀仪,真空度为1×10-6mbar,蒸镀速率设为0.1nm s-1,在玻璃或塑料衬底上蒸镀材料为氧化铟锡,厚度为100nm的阳极导电层;
二、在阳极导电层上蒸镀材料为MoO3,厚度为20nm的空穴注入层;
三、在空穴注入层上蒸镀材料为NPB,厚度为50nm的空穴传输层;
四、在空穴传输层上蒸镀材料为mCP,厚度为10nm的电子阻挡层;
五、在电子阻挡层上继续蒸镀厚度为30nm的主体材料(基于膦杂芳基衍生物的热激发延迟荧光主体材料)和客体材料4CzTPNPh(客体材料的掺杂浓度为14%)的发光层;
六、在发光层上蒸镀材料为DPEPO,厚度为15nm的空穴阻挡层;
七、在空穴阻挡层上蒸镀材料为Bphen,厚度为40nm的电子传输层;
八、在电子传输层上蒸镀材料为LiF,厚度为20nm的电子注入层;
九、在电子注入层上蒸镀材料为金属Al,厚度为150nm的阴极导电层,封装,得到热激发延迟荧光(TADF)电致发光器件。
本应用实施例制备的电致发光器件的电压-电流密度关系曲线如图27所示。其中的□表示4tCzDPDPA,○表示4tCzDPDPSA,△表示4tCzDPDPS2A,▽表示4tCzDPDPSOA,◇表示4tCzDPDPO2A。从图27可以看出,随着电压的升高,器件的电流密度都呈现出升高的趋势。
本应用实施例的电致发光器件的电压-亮度关系曲线如图28所示,其中其中的□表示4tCzDPDPA,○表示4tCzDPDPSA,△表示4tCzDPDPS2A,▽表示4tCzDPDPSOA,◇表示4tCzDPDPO2A。由此图可知以4tCzDPDPA制备的电致发光器件的启亮电压为3.5V,以4tCzDPDPSA制备的电致发光器件的启亮电压为3.5V,以4tCzDPDPS2A制备的电致发光器件的启亮电压为3.6V,以4tCzDPDPSOA制备的电致发光器件的启亮电压为3.6V,以4CzDPDPO2A制备的电致发光器件的启亮电压为3.6V。
本应用实施例的电致发光器件的亮度-电流效率关系曲线如图29所示,其中的□表示4tCzDPDPA,○表示4tCzDPDPSA,△表示4tCzDPDPS2A,▽表示4tCzDPDPSOA,◇表示4tCzDPDPO2A。从图中可以看出,以4tCzDPDPA制备的电致发光器件的电流效率达到最大值为7.4cd·A-1,以4tCzDPDPSA制备的电致发光器件的电流效率达到最大值为8.2cd·A-1,以4tCzDPDPS2A制备的电致发光器件的电流效率达到最大值为8.4cd·A-1,以4tCzDPDPSOA制备的电致发光器件的电流效率达到最大值为7.6cd·A-1,以4tCz DPDPO2A制备的电致发光器件的电流效率达到最大值为9.4cd·A-1。
本应用实施例的电致发光器件的亮度-功率效率关系曲线如图30所示,其中的□表示4tCzDPDPA,○表示4tCzDPDPSA,△表示4tCzDPDPS2A,▽表示4tCzDPDPSOA,◇表示4tCzDPDPO2A。由此图可知以4tCz DPDPA制备的电致发光器件的功率效率达到最大值为4.2lm·W-1,以4tCzDPDPSA制备的电致发光器件的功率效率达到最大值为5.6lm·W-1,以4tCzDPDPS2A制备的电致发光器件的功率效率达到最大值为6.6lm·W-1,以4tCzDPDPSOA制备的电致发光器件的功率效率达到最大值为3.6lm·W-1,以4tCzDPDPO2A制备的电致发光器件的功率效率达到最大值为7.9lm·W-1。
本应用实施例的电致发光器件的亮度-外量子效率关系曲线如图31所示,其中□表示4tCzDPDPA,○表示4tCz DPDPSA,△表示4tCz DPDPS2A,▽表示4tCz DPDPSOA,◇表示4tCz DPDPO2A,由此图可知以4tCzDPDPA制备的电致发光器件的最大外量子效率4.2%,以4tCzDPDPSA制备的电致发光器件的最大外量子效率4.5%,以4tCzDPDPS2A制备的电致发光器件的最大外量子效率4.6%,以4tCzDPDPSOA制备的电致发光器件的最大外量子效率4.1%,以4tCzDPDPO2A制备的电致发光器件的的最大外量子效率5.1%。
本应用实施例的电致发光器件的电致发光光谱图如图32所示,□表示4tCzDPDPA,○表示4tCzDPDPSA,△表示4tCzDPDPS2A,▽表示4tCzDPDPSOA,◇表示4tCzDPDPO2A,由此图可知以4tCzDPDPA制备的电致发光器件的电致发光峰在596nm处、以4tCzDPDPSA制备的电致发光器件的电致发光峰在590nm处、以4tCzDPDPS2A制备的电致发光器件的电致发光峰在590nm处、以4tCzDPDPSOA制备的电致发光器件的电致发光峰在597nm处,以4tCzDPDPO2A制备的电致发光器件的电致发光峰在598nm处。
实施例十一:本实施方式本实施例的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzPPDPO的合成方法按下列步骤进行:
将1mmol1,2-二溴-4,5-二咔唑苯、1.2mmol的苯基二氯化膦、2mmol正丁基锂和20ml的THF混合于50ml三颈圆底烧瓶中,在氩气保护下,在温度为-78℃下搅拌3小时;加入1.5mmol二苯基氯化膦,在温度为78℃的条件下反应3小时,然后倒入水中,用二氯甲烷萃取,得到有机层,向有机层中加入12mmolH2O2,在温度为0℃的条件下搅拌进行氧化反应0.5小时;用亚硫酸氢钠溶液和水分别洗涤,二氯甲烷萃取,无水硫酸钠干燥后,旋转蒸发除去二氯甲烷溶剂,以乙醇和二氯甲烷体积比1:20的混合溶剂为淋洗剂柱层析纯化,得到4CzPPDPO。
本实施例制备的4CzPPDPO,质谱仪测得数据为:m/z:1138.36(100.0%),1139.36(85.0%),1140.36(36.9%),1141.37(9.9%),1142.37(2.2%),1139.35(1.5%);GC-MS:m/z(%):1139(100)[M+];Elemental Analysis of C78H52N4O2P2:C,82.23;H,4.60;N,4.92;O,2.81。从而可知4CzPPDPO的结构式为:
本实施例得到基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4CzPPDPO的紫外荧光光谱谱图如图33所示。
本实施例得到基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4CzPPDPO的热重分析谱图如图34所示,由图可知本实施例基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4CzPPDPO的裂解温度达372℃。
实施例十二:本实施例的基于膦杂芳基衍生物的热激发延迟荧光主体材料4CzPPTPO的合成方法按下列步骤实现:
将1mmol 1,2-二溴-4,5-二叔丁基咔唑苯、1.2mmol的苯基二氯化膦、2.2mmol正丁基锂加入到装有25mlTHF的50ml三颈圆底烧瓶中,在氩气保护下,在温度为-78℃下搅拌3小时后,再加入2.3mmol的二苯基氯化膦,反应2小时后倒入水中,再用二氯甲烷萃取,得到有机层,加入25mmol的H2O2,在温度为0℃的条件下搅拌进行氧化反应1小时;用亚硫酸氢钠溶液和水分别洗涤,再用二氯甲烷萃取,无水硫酸钠干燥后,旋转蒸发除去二氯甲烷溶剂,以乙醇和二氯甲烷体积比1:20的混合溶剂为淋洗剂柱层析纯化,得到4CzPPTPO。
本实施例制备的4CzPPTPO,质谱仪测得数据为:m/z:1338.40(100.0%),1339.40(98.2%),1340.40(49.0%),1341.41(15.3%),1342.41(3.9%),1339.39(1.5%),1341.40(1.3%);GC-MS:m/z(%):1339(100)[M+];Elemental Analysis of C90H61N4O3P3:C,80.71;H,4.59;N,4.18;O,3.58。从而可知4CzPPTPO的结构式为
本实施例得到基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4CzPPTPO的紫外荧光光谱谱图如图35所示。
本实施例得到基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4CzPPTPO的热重分析谱图如图36所示,由图可知本实施例基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4CzPPTPO的裂解温度达372℃。
实施例十三:本实施例基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4tCzPPDPO的合成方法按下列步骤实现:
将1mmol 1,2-二溴-4,5-二叔丁基咔唑苯、1.5mmol的苯基二氯化膦、2.1mmol正丁基锂和25ml的THF混合于50ml三颈圆底烧瓶中,在氩气保护下,在温度为-78℃下搅拌3小时后,再加入1.3mmol的二苯基氯化膦,反应2小时后倒入水中,再用二氯甲烷萃取,得到有机层,向有机层液体中加入25mmol的12mmolH2O2,在温度为0℃的条件下搅拌进行氧化反应1小时;用亚硫酸氢钠溶液和水分别洗涤,再用二氯甲烷萃取,无水硫酸钠干燥后,、旋转蒸发去除二氯甲烷溶剂,以乙醇和二氯甲烷体积比1:20的混合溶剂为淋洗剂柱层析纯化,得到4tCzPPDPO。
本实施例制备的4tCzPPDPO,质谱仪测得数据为:m/z:1587.86(100.0%),1586.86(83.1%),1588.86(60.1%),1589.87(23.5%),1590.87(7.1%),1591.87(1.7%),1588.87(1.3%),1589.86(1.3%),1587.85(1.2%);GC-MS:m/z(%):1588(100)[M+];Elemental Analysis of C110H116N4O2P2:C,83.19;H,7.36;N,3.53;O,2.01。从而可知4tCzPPDPO的结构式为
本实施例得到基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4tCzPPDPO的紫外荧光光谱谱图如图37所示。
本实施例得到基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4tCzPPDPO的热重分析谱图如图38所示,由图可知本实施例基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4tCzPPDPO的裂解温度达372℃。
实施例十四:本实施例基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4tCzPPTPO的合成方法按下列步骤实现:
将1mmol 1,2-二溴-4,5-二叔丁基咔唑苯、1.5mmol的苯基二氯化膦、2.1mmol正丁基锂和25ml的THF混合于50ml三颈圆底烧瓶中,在氩气保护下,在温度为-78℃下搅拌3小时后,加入2.8mmol的二苯基氯化膦,反应2小时后,倒入水中,再用二氯甲烷萃取,得到有机层,向有机层中加入30mmol的H2O2,在温度为0℃的条件下搅拌进行氧化反应1小时;用亚硫酸氢钠溶液和水分别洗涤,二氯甲烷萃取,无水硫酸钠干燥后,旋转蒸发去除二氯甲烷溶剂,以乙醇和二氯甲烷体积比1:20的混合溶剂为淋洗剂柱层析纯化,得到4tCzPPTPO。
本实施例制备的4tCzPPTPO,质谱仪测得数据为:m/z:1787.90(100.0%),1786.90(74.9%),1788.90(66.7%),1789.91(29.0%),1790.91(9.9%),1791.91(2.6%),1789.90(1.6%),1788.91(1.4%),1787.89(1.1%);GC-MS:m/z(%):1788(100)[M+];Elemental Analysis of C122H125N4O3P3:C,81.94;H,7.05;N,3.13;O,2.68。从而可知4tCzPPTPO的结构式为
本实施例得到基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4tCzPPTPO的紫外荧光光谱谱图如图39所示。
本实施例得到基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4tCzPPTPO的热重分析谱图如图40所示,由图可知本实施例基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4tCzPPTPO的裂解温度达372℃。
应用实施例三:利用实施例十一至十四制备的基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料4CzPPDPO、4CzPPTPO、4tCzPPDPO、4tCzPPTPO制备电致发光器件的方法,按以下步骤进行:
一、将经过去离子水清洗的玻璃或塑料衬底放入真空蒸镀仪,真空度为1×10-6mbar,蒸镀速率设为0.1nm s-1,在玻璃或塑料衬底上蒸镀材料为氧化铟锡,厚度为100nm的阳极导电层;
二、在阳极导电层上蒸镀材料为MoO3,厚度为20nm的空穴注入层;
三、在空穴注入层上蒸镀材料为NPB,厚度为45nm的空穴传输层;
四、在空穴传输层上蒸镀材料为mCP,厚度为10nm的电子阻挡层;
五、在电子阻挡层上继续蒸镀厚度为30nm主体(基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料)和客体(TADF客体材料4CzTPNPh,客体的掺杂浓度为5%~15%);
六、在发光层上蒸镀材料为DPEPO,厚度为10nm的空穴阻挡层;
七、在空穴阻挡层上蒸镀材料为BPhen,厚度为30nm的电子传输层;
八、在电子传输层上蒸镀材料为LiF,厚度为20nm的电子注入层;
九、在电子注入层上蒸镀材料为金属Al,厚度为150nm的阴极导电层,封装,得到热激发延迟荧光(TADF)电致发光器件。
本应用实施例以基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料制备的电致发光器件的电压-电流密度关系曲线如图41所示,其中的■表示4CzPPDPO、●表示4CzPPTPO、▲表示4tCzPPDPO、▼4tCzPPTPO表示。
本实施例以基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料制备的电致发光器件的电压-亮度关系曲线如图42所示,其中的■表示4CzPPDPO、●表示4CzPPTPO、▲表示4tCzPPDPO、▼表示4tCzPPTPO,从图42可以看出,以4CzPPDPO制备的电致发光器件的启亮电压为4.1V,以4CzPPTPO制备的电致发光器件的启亮电压为4.2V,以4tCzPPDPO制备的电致发光器件的启亮电压为4.1V,以4tCzPPTPO制备的电致发光器件的启亮电压为4.2V。
本实施例以基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料制备的电致发光器件的亮度-电流效率关系曲线如图43所示,其中的■表示4CzPPDPO、●表示4CzPPTPO、▲表示4tCzPPDPO、▼表示4tCzPPTPO,由此图可知,以4CzPPDPO制备的电致发光器件的电流效率达到最大值20.6cd·A-1,以4CzPPTPO制备的电致发光器件的的电流效率达到最大值16.5cd·A-1,以4tCzPPDPO制备的电致发光器件的的电流效率达到最大值20cd·A-1,以4tCzPPTPO制备的电致发光器件的的电流效率达到最大值9.1cd·A-1。
本实施例以基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料制备的电致发光器件的亮度-功率效率关系曲线如图44所示,其中的■表示4CzPPDPO、●表示4CzPPTPO、▲表示4tCzPPDPO、▼表示4tCzPPTPO,由此图可知以4CzPPDPO制备的电致发光器件的功率效率达到最大值14.7lm·W-1,以4CzPPTPO制备的电致发光器件的功率效率达到最大值10.3lm·W-1,以4tCzPPDPO制备的电致发光器件功率效率达到最大值14.4lm·W-1,,以4tCzPPTPO制备的电致发光器件的功率效率达到最大值5.7lm·W-1。
本实施例以基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料制备的电致发光器件的亮度-功率效率关系曲线如图45所示,其中的■表示4CzPPDPO、●表示4CzPPTPO、▲表示4tCzPPDPO、▼表示4tCzPPTPO。由此图可知以4CzPPDPO制备的电致发光器件的最大外量子效率11.5%,以4CzPPTPO制备的电致发光器件的的最大外量子效率9.4%,以4tCzPPDPO制备的电致发光器件的最大外量子效率11.1%,以4tCzPPTPO制备的电致发光器件的最大外量子效率5.1%。
本实施例以基于膦杂芳基衍生物的热激发延迟荧光器件的主体材料制备的电致发光器件的电致发光光谱图如图46所示,其中■表示4CzPPDPO、●表示4CzPPTPO、▲表示4tCzPPDPO、▼表示4tCzPPTPO,由此图可知4CzPPDPO制备的电致发光器件的电致发光峰在615nm处,以4CzPPTPO制备的电致发光器件的电致发光峰在596nm处,以4tCzPPDPO制备的电致发光器件的电致发光峰在52nm处以4tCzPPTPO制备的电致发光器件的电致发光峰在582nm处。
- 还没有人留言评论。精彩留言会获得点赞!