一种无乳链球菌的检测引物组、检测试剂盒和多重PCR检测方法与流程
本发明属于水产养殖动物病原菌检测领域,具体涉及一种无乳链球菌的检测引物组。同时本发明还涉及了包含上述引物组的检测试剂盒以及使用所述试剂盒的多重PCR检测方法。
背景技术:
无乳链球菌(Streptococcus agalactiae)隶属于芽孢杆菌纲,乳杆菌目,链球菌科,链球菌属,为兼性革兰氏阳性球菌,是养殖鱼类的重要病原菌之一,无乳链球菌还能感染人、哺乳动物,是重要的人兽鱼共患病原菌。至今为止,发现有30 多种养殖淡水鱼类和50 多种海水鱼类容易受到无乳链球菌的感染,世界水产业每年因无乳链球菌病造成的损失均高达100 亿美元,其中以罗非鱼最易受到该病菌的入侵。无乳链球菌病发病较快,除一部分急性死亡外,并没有明显的症状,因此从发病鱼的症状上很难判断病原菌的种类,从而对疾病的有效处理产生影响。因此,在水产养殖生产中建立对无乳链球菌的快速检测技术,是有效预防链球菌病的重要手段,也为提高水产品质量安全提供保障。目前,关于无乳链球菌检测主要依靠病原菌的分离和生理生化鉴定等方法,这些方法操作繁琐、检测周期长,不能达到快速鉴定的目的。分子鉴定具有快速、简便以及灵敏度高的优点,是微生物鉴定的发展趋势。虽然有关无乳链球菌的分子检测方法国内外已有报道,但多数是利用纯培养的菌液进行检测,使用普通PCR方法,一次只能检测一个基因,常常会造成漏检或误检。
多重PCR是在一次反应中能同时扩增多个目的片段的靶序列,该技术可用于多种病原微生物的检测或鉴定,在同一PCR反应管中同时加多对特异性引物,进行PCR扩增,其最大的问题在于引物的设计和反应条件的优化,因为各种引物混合后扩增会有条带丢失现象,各引物间还存在竞争关系,强势引物可能掩盖弱势引物,导致目的片段丢失,甚至引物间相互作用会产生严重的引物二聚体。目前有构建无乳链球菌三重PCR检测方法的文献报道,但该检测技术对无乳链球菌DNA 样本的最低检测浓度仅为3.2×10-1 ng/μL。有鉴于此,开发一种快速、高效、特异性好、灵敏度高的无乳链球菌多重PCR势在必行,这对于加强水产品养殖水体、水产品中无乳链球菌的快速检验检疫、水产养殖病害的早期监测和预警、无公害水产品检测和生产,以及水产品卫生质量监督检验等方面具有重要的意义。
技术实现要素:
针对上述不足,本发明的目的之一是提供一种检测无乳链球菌的检测引物组,该引物组对无乳链球菌的特异性好、灵敏度高。
编码无乳链球菌透明质酸裂解酶的hylB 基因、青霉素结合蛋白ponA 基因和成孔毒素特性CAMP因子的cfb 基因是真正与致病相关的无乳链球毒力基因,以它们为靶标设计引物,可以提高检测的特异性和灵敏度。故,本发明的第一个目的通过以下技术方案来实现:
一种无乳链球菌的检测引物组,包括引物对hylB、引物对ponA和引物对cfb;其中,所述引物对hylB 包括核苷酸序列如SEQ ID NO.1所示的引物hylB-F和核苷酸序列如SEQ ID NO.2所示的引物hylB-R;所述引物对ponA包括核苷酸序列如SEQ ID NO.3所示的引物ponA-F和核苷酸序列如SEQ ID NO.4所示的引物ponA-R;所述引物对cfb包括核苷酸序列如SEQ ID NO.5所示的引物cfb-F和核苷酸序列如SEQ ID NO.6所示的引物cfb-R。
本发明目的之二是提供含有上述的无乳链球菌检测引物组的试剂盒。
进一步地,所述的无乳链球菌的检测试剂盒,还包含TE缓冲液、溶菌酶、蛋白酶K、SDS溶液、酚-氯仿-异戊醇混合溶液、异丙醇、体积浓度为70%的乙醇、CTAB的NaCl溶液、阳性对照品和PCR DsMix。
进一步地,上述的无乳链球菌的检测试剂盒,所述酚-氯仿-异戊醇混合溶液中酚、氯仿和异戊醇的体积比为26~24:23.2~24.8:0.8~1.2。
进一步地,上述的无乳链球菌的检测试剂盒,所述CTAB的NaCl溶液为将CTAB溶于0.5 mol/L的NaCl溶液中,CTAB和所述NaCl溶液的质量体积比为0.8~1.2:20.2~19.8。
进一步地,上述的无乳链球菌的检测试剂盒,所述阳性对照品为无乳链球菌的DNA模板。
进一步地,上述无乳链球菌的检测试剂盒,所述检测引物组中的引物浓度均为10 μM。
本发明目的之三是提供使用上述试剂盒检测无乳链球菌的多重PCR检测方法。
使用上述试剂盒检测无乳链球菌的多重PCR检测方法,包括如下步骤:
1)取待检测样品50~100 mg并加入1000 μL的TE缓冲液,用匀浆器充分匀浆,取组织匀浆液180 μL,转入到1.5ml的离心管中,加入20 μL 浓度为50 mg/mL的溶菌酶,30℃孵育10 min;
2)向步骤1)溶液中加入10 μL质量浓度为10%SDS和5 μL 20mg/mL的蛋白酶K,并于37℃温育1h;
3)向步骤2)温育后的溶液中加入50 μL 5 mol/L的NaCl溶液, 充分混匀后再加入40 μL CTAB的NaCl溶液,65℃温育20 min;
4)向步骤3)温育后的溶液中加入与温育后的溶液等体积的酚-氯仿-异戊醇混合溶液混匀,12000 g/min 离心4-5 min;
5)将步骤4)离心后的上清转入一只新管中,加入上清液0.6-0.8倍体积的异丙醇,12000 g/min离心4-5 min;
6)去上清,沉淀用1 mL的体积浓度为70%的乙醇洗涤后,12000 g/min离心4-5 min;
7)步骤6)离心后去上清,沉淀常温干燥5-10 min,重溶于30-50 μL TE缓冲液,即为样品DNA模板;
8)配制样品组多重PCR反应体系包括:12.5 μL 2×PCR DsMix、0.5 μL引物hylB-F、0.5 μL引物hylB-R、0.6 μL引物ponA-F、0.6 μL引物ponA-R、0.5 μL引物cfb-F、0.5 μL引物cfb-R、1.0 μL步骤7)得到的样品DNA模板和8.3 μL无菌水;
9)配制阳性对照组多重PCR反应体系包括:12.5 μL 2×PCR DsMix、0.5 μL引物hylB-F、0.5 μL引物hylB-R、0.6 μL引物ponA-F、0.6 μL引物ponA-R、0.5 μL引物cfb-F、0.5 μL引物cfb-R、1.0 μL阳性对照品和8.3 μL无菌水;
10)将步骤8)制得的样品组和步骤9)制得的阳性对照组多重PCR反应体系分别混匀后12000 g/min 离心10 s,置于PCR仪中,分别于94℃预变性4 min,94℃变性30 s、57℃退火30 s、72℃延伸1 min,30个循环,72℃延伸10 min,进行PCR反应;
11)对步骤10)PCR反应结果进行电泳检测,并于凝胶成像系统下观察结果,若样品孔电泳条带出现与阳性对照孔相同大小的条带,则说明待测样品中含有无乳链球菌。
所述步骤(11) 电泳检测中,分别设置样品孔、阳性对照孔和参比孔;所述样品孔取步骤10)样品组PCR反应产物5 μL与6×Loading Buffer 1μL混合,加入到1%琼脂糖凝胶点样孔中;所述阳性对照孔取步骤10)阳性对照组PCR反应产物5 μL与6×Loading Buffer 1 μL混合,加入到1%琼脂糖凝胶点样孔中;所述参比孔取5 μL DL2000 DNA Marker加入到1%琼脂糖凝胶点样孔中;以120 V电压分别对所述样品孔、所述阳性对照孔和所述参比孔进行电泳25 min。
本发明上述检测方法可用于检测包括动物体、水产品、养殖环境、养殖用食物、等是否存在无乳链球菌。所述养殖环境包括养殖水体和养殖土壤等。
相比现有技术,本发明具有如下有益效果:
1.本发明引物组具有高特异性,可以根据是否扩增就能判断目标基因的存在与否,从而能够确定样品是否含有无乳链球菌,能够实现对该菌进行检测。
2.采用本发明检测试剂盒可以同时对无乳链球菌三种重要的毒力基因进行检测,大大提高了检测的灵敏度,检测无乳链球菌DNA的最低浓度为7.74×10-3 ng/ uL,比文献报道的无乳链球菌多重PCR快速检测方法敏感性提高200倍以上,并且在5个小时左右完成检测。相比传统检测方法,不仅节约了成本和时间,也减少了劳动力的支出。
3.本发明的无乳链球菌多重PCR检测方法,不但使得养殖鱼体内和环境中病原检测更加快速,而且可用于鱼类养殖过程中各时期跟踪检测中,可以对病害的爆发进行及时预警,避免病原菌传播流行,降低养殖鱼类疾病爆发的风险,提高水产品质量安全,具有很高的实用价值,有利于增加水产养殖的经济和社会效益。
4.本发明的检测操作简单,不需要使用复杂仪器,也不需要特殊试剂,只需常规PCR仪即可,对检测人员的技术素质要求较低,只需进行简单培训即可。
5.本发明提供的引物组及检测方法特异性好、灵敏度高、简单快速、高效精准,适用于无公害水产品中无乳链球菌的快速检验检疫,可直接应用于水产养殖病害的早期监测和预警。
6. 本发明提供的引物组及检测方法应用范围广泛,不仅可以检测鱼类体内和养殖水体中是否存在的无乳链球菌,也可针对其他环境或动物中是否存在无乳链球菌进行快速检测。
7.本发明检测试剂盒制备成本低廉,制备工艺简单,不需经过细菌培养,检测所需样品量少,可实现微创取样,容易实现工业化大生产,具有广阔的市场前景。
附图说明
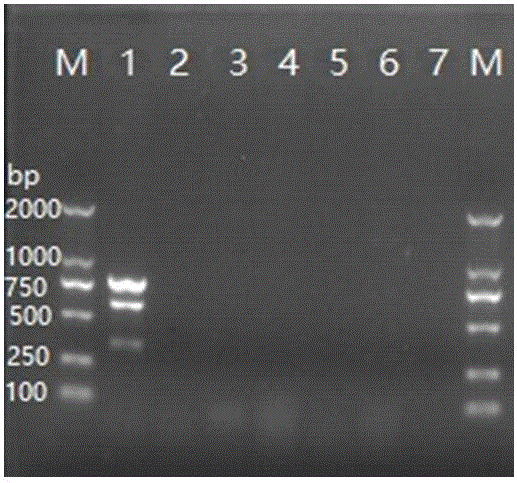
图1为实施例2中多重PCR扩增产物及单一PCR扩增产物检测结果电泳图。M为Marker DL2000;泳道1为多重PCR的扩增结果,从上到下依次为hylB、ponA和cfb基因;泳道2为hylB基因单一PCR的扩增结果;泳道3为ponA基因单一PCR的扩增结果;泳道4为cfb基因单一PCR的扩增结果;泳道5为阴性对照。
图2为实施例5中多重PCR特异性试验。M为Marker DL2000;泳道1为无乳链球菌DNA模板;泳道2为海豚链球菌DNA模板;泳道3为美人鱼发光杆菌DNA模板;泳道4为粪肠球菌DNA模板;泳道5为葡萄球菌DNA模板;泳道6为迟缓爱德华菌DNA模板;泳道7为无菌水。
图3为实施例6中多重PCR灵敏性试验。M为Marker DL2000;泳道1为无乳链球菌DNA浓度2.42×101 ng/uL;泳道2为无乳链球菌DNA浓度4.84×100 ng/uL;泳道3为无乳链球菌DNA浓度9.68×10-1 ng/ uL;泳道4为无乳链球菌DNA浓度1.94×10-1 ng/ uL;泳道5为无乳链球菌DNA浓度3.87×10-2 ng/ uL;泳道6为无乳链球菌DNA浓度7.74×10-3 ng/ uL;泳道7为无乳链球菌DNA浓度1.55×10-3 ng/ uL。
具体实施方式
下面结合具体实施例和附图对本发明作进一步详细说明。本实施案例在以本发明技术为前提下进行实施,现给出详细的实施方式和具体的操作过程,但本发明的保护范围不限于以下的实施例。
实施例1 样品基因组DNA的提取
1. 鱼(包括鱼体内细菌)基因组DNA提取:
1)取鱼组织待检测样品50~100 mg并加入1000 μL的TE缓冲液,用匀浆器充分匀浆,取组织匀浆液180 μL,转入到1.5ml的离心管中,加入20 μL 浓度为50 mg/mL的溶菌酶,30 ℃孵育10 min。
2)向步骤1)溶液中加入10 μL质量浓度为10%SDS和5 μL 20mg/mL的蛋白酶K,并于37℃温育1h。
3)向步骤2)温育后的溶液中加入50 μL 5 mol/L的NaCl溶液, 充分混匀后再加入40 μL CTAB的NaCl溶液,65℃温育20 min。
4)向步骤3)温育后的溶液中加入与温育后的溶液等体积的酚-氯仿-异戊醇混合溶液混匀,12000 g/min 离心4-5 min。
5)将步骤4)离心后的上清转入一只新管中,加入上清液0.6-0.8倍体积的异丙醇,12000 g/min离心4-5 min。
6)步骤5)离心后去上清,沉淀用1 mL的体积浓度为70%的乙醇洗涤后,12000 g/min离心4-5 min。
7)步骤6)离心后去上清,沉淀常温干燥5-10 min,重溶于30-50 μL TE缓冲液,即为样品DNA模板。
2. 细菌基因组DNA提取:
1)分别离心收集培养的无乳链球菌、海豚链球菌、粪肠球菌、美人鱼发光杆菌、葡萄球菌和迟缓爱德华氏菌,分别用1000 μL的TE缓冲液重悬细菌,取细菌重悬液180 μL,转入到1.5ml的离心管中,加入20 μL 浓度为50 mg/mL的溶菌酶,30 ℃孵育10 min。
2)向步骤1)溶液中加入10 μL质量浓度为10%SDS和5 μL 20mg/mL的蛋白酶K,并于37℃温育1h。
3)向步骤2)温育后的溶液中加入50 μL 5 mol/L的NaCl溶液, 充分混匀后再加入40 μL CTAB的NaCl溶液,65℃温育20 min。
4)向步骤3)温育后的溶液中加入与温育后的溶液等体积的酚-氯仿-异戊醇混合溶液混匀,12000 g/min 离心4-5 min。
5)将步骤4)离心后的上清转入一只新管中,加入上清液0.6-0.8倍体积的异丙醇,12000 g/min离心4-5 min。
6)步骤5)离心后去上清,沉淀用1 mL的体积浓度为70%的乙醇洗涤后,12000 g/min离心4-5 min。
7)步骤6)离心后去上清,沉淀常温干燥5-10 min,重溶于30-50 μL TE缓冲液,即为细菌基因组DNA模板。
实施例2 无乳链球菌多重PCR检测引物组的设计和有效性检测
1.选取无乳链球菌三个的特异性毒力基因hylB、ponA和cfb,采用Primer Premier 6.0软件分析设计出相应的引物对,各引物对能分别各自专一性鉴别上述细菌毒力基因的特异性,引物序列如下所示:
2.有效性检测:
(1)合成上述设计的引物组,用引物组的引物分别对实施例1提取的无乳链球菌的DNA进行单一PCR验证,其中单一PCR反应体系如下:
(2)同时对无乳链球菌三个毒力基因进行多重PCR验证,采用如下多重PCR反应体系:
(3)采用如下PCR反应程序,对上述PCR反应体系进行PCR反应:
1)94℃预变性4 min。
2)94℃变性30 s。
57℃退火30 s。
72℃延伸1 min。
共计30个循环。
3)72℃延伸10 min。
上述PCR反应结束后,分别取各组PCR反应产物5 μL与6×Loading Buffer 1 μL混合,分别加入到质量浓度为1%的琼脂糖凝胶点样孔中,同时在样品孔旁添加对照组,对照组琼脂糖凝胶点样孔中加入5 μL DL2000 DNA Marker作对照,以120V电压进行电泳,约25 min后于凝胶成像系统下观察结果。
3.检测结果:
检测结果如图1所示,图1中泳道M为Marker DL2000;泳道1为无乳链球菌三个毒力基因的多重PCR的扩增结果,从上到下依次为hylB、ponA和cfb基因;泳道2为hylB基因单一PCR的扩增结果,目的扩增片段长度为790 bp;泳道3为ponA基因单一PCR的扩增结果,目的扩增片段长度为598 bp;泳道4为cfb基因单一PCR的扩增结果,目的扩增片段长度为348 bp;泳道5为三个毒力基因多重PCR的阴性对照;从图1可以看出单一PCR检测相应的单一条带及多重PCR检测相应的3条单一条带,分别与hylB、ponA和cfb基因的扩增片段长度分别为790、598和348 bp相吻合,即说明本发明的无乳链球菌检测用引物组可以准确、特异性地检测出无乳链球菌三种重要的毒力基因。
实施例4 试剂盒
本实施例的检测试剂盒可用于多重PCR快速检测样品中是否含有无乳链球菌,该检测试剂盒由无乳链球菌的hylB、ponA和cfb基因检测用引物组、TE缓冲液、溶菌酶、蛋白酶K、SDS溶液、酚-氯仿-异戊醇混合溶液、异丙醇、体积浓度为70%的乙醇、CTAB的NaCl溶液、阳性对照品和PCR DsMix组成,其中:
(1)无乳链球菌检测用引物组:1管内装无乳链球菌的hylB基因引物hylB-F及hylB-R,其核苷酸序列分别如SEQ ID NO.1和SEQ ID NO.2所示;2管内装无乳链球菌的ponA基因引物ponA-F及ponA-R,其核苷酸序列分别如SEQ ID NO.3和SEQ ID NO.4所示;3管内装无乳链球菌的cfb基因引物cfb-F及cfb-R,其核苷酸序列分别如SEQ ID NO.5和SEQ ID NO.6所示;以上引物浓度分别为10 μM。
(2)溶菌酶(50 mg/mL ),置于容器,1管。
(3)蛋白酶K(20 mg/mL),置于容器,1管。
(4)制备TE缓冲液(10 mM Tris-HCl, 0.1 mM EDTA,pH8.0),置于容器,1管。
(5)质量浓度为10% SDS,置于容器,1管。
(6)酚-氯仿-异戊醇混合溶液,酚-氯仿-异戊醇体积比25:24:1,置于容器,1管。
(7)异丙醇,置于容器,1管。
(8)制备体积浓度为70%乙醇,置于容器,1管。
(9)制备CTAB的NaCl溶液,5 g CTAB溶于100 mL 0.5 M NaCl溶液中,置于容器,1管。
(10)制备阳性对照品:提取无乳链球菌的基因组DNA,置于容器,1管。
(11)2×PCR DsMix,置于容器,1管。
(12)一块泡沫板,将上述13个小管分别对应放置于泡沫板的孔内,装于盒中即制备得到本实施例的检测试剂盒。
本实施例中各试剂管制备后均经过抽样质检,对其质量进行监控。
实施例5 利用实施例4制得的试剂盒进行多重PCR特异性试验
分别准备以下样品:样品1含有无乳链球菌,样品2含有海豚链球菌,样品3含有美人鱼发光杆菌,样品4含有粪肠球菌,样品5含有葡萄球菌,样品6含有迟缓爱德华菌,样品7为无菌水。分别以实施例1的方法提取上述各样品中的DNA作为多重PCR的模板,以实施例2中的方法对各样品进行多重PCR检测。
检测结果如图2所示,从图中可看出本发明的三重PCR检测方法可以同时检测到无乳链球菌的三个特异的毒力基因;不受其他水生动物常见病原菌的干扰,本发明建立的多重PCR检测方法特异性强。
实施例6 利用实施例4制得的试剂盒进行多重PCR灵敏性试验
取28℃培养20h的无乳链球菌菌液,以实施例1的方法提取细菌基因组DNA,对基因组DNA进行5倍的系列梯度稀释,以上述浓度为2.42×101 ng/uL、4.84×100 ng/uL、9.68×10-1 ng/ uL、1.94×10-1 ng/ uL、3.87×10-2 ng/ uL、7.74×10-3 ng/ uL和1.55×10-3 ng/ uL的基因组DNA为模板,以实施例2中的方法对各样品进行多重PCR检测。
检测结果如图3所示,本发明的检测方法对含有无乳链球菌基因组DNA浓度为 7.74×10-3 ng/ uL的样品仍能检出,可见本发明的多重PCR检测无乳链球菌的检测限可达7.74×10-3 ng/ uL,该检测方法具有较高的灵敏性,比文献报道的无乳链球菌多重PCR检测方法提高了200倍。
本发明可用其他的不违背本发明的精神或主要特征的具体形式来概述。本发明的上述实施例都只能认为是对本发明的说明而不是限制。如可采用上述实施例的引物组和检测方法对养殖水体、养殖土壤、养殖食物等进行检测,以判断是否存在无乳链球菌。因此凡是依据本发明的实质技术对以上实施例所作的任何细微修改、等同变化与修饰,均属于本发明技术方案的范围内。
序列表
<110> 中国水产科学研究院南海水产研究所
<120>一种无乳链球菌的检测引物组、检测试剂盒和多重PCR检测方法
<160> 6
<210> 1
<211> 20
<212> DNA
<213> 引物hylB-F
<400> 1
CGCGACTTTG GCTTTCTGAG 20
<210> 2
<211> 20
<212> DNA
<213> 引物hylB-R
<400> 2
TAATTGAGCG AGGGACACCG 20
<210> 3
<211> 20
<212> DNA
<213> 引物ponA-F
<400> 3
AGCTATCCCT GGTGTTGCAC 20
<210> 4
<211> 20
<212> DNA
<213> 引物ponA-R
<400> 4
ACCGTTAGGT ACTGTATTGT TGT 23
<210> 5
<211> 20
<212> DNA
<213> 引物cfb-F
<400> 5
TGGGATTTGG GATAACTAAG C 21
<210> 6
<211> 20
<212> DNA
<213> 引物cfb-R
<400> 6
TTTGAAGTGC TGCTTGTAAT G 21
- 还没有人留言评论。精彩留言会获得点赞!