一种喜树碱类前药及其制备和应用的制作方法
本发明属于药物控制释放技术领域,尤其涉及一种喜树碱类和伊文思蓝形成的小分子两亲性药物及其制备方法和应用。
背景技术:
癌症是全球发病和死亡的主要原因之一。根据世界卫生组织统计,2012年全球约有1400万新病例,并预计在未来二十年新病例的数量将增加约70%。癌症是全球第二大死亡原因,2015年造成880万人死亡。全球,接近六分之一的死亡是由于癌症,其中约70%的癌症死亡发生在低收入和中等收入国家。目前,化疗是癌症的主要治疗方法之一。然而,大多数化疗药物,比如喜树碱(camptothecin),紫杉醇(paclitaxel),阿霉素(doxorubicin)等,在水中溶解度都很底,而且有着不良副作用。例如,喜树碱在临床前研究中具有显著的抗肿瘤活性,但由于低溶解度和严重不良副作用,在临床试验中失败。目前,喜树碱类似物伊立替康(irinotecan)和拓扑替康(topotecan)已被美国食品和药物管理局(fda)批准用于治疗结肠癌。伊立替康显示出更好的水溶性和减少的副作用;然而,对癌细胞的杀伤力也明显受损。
纳米药物已被广泛研究用于癌症治疗。纳米颗粒的使用不仅可以改善这些药物的水分散性,而且还可以增强它们的药代动力学和生物体内分布,改善治疗效果和减少副作用。小分子两亲性前药组装成的纳米颗粒受到广泛研究。小分子两亲性药物,一般由疏水性的化疗药物连接到亲水性小分子,例如低聚乙二醇,多肽序列,或另一种亲水性药物。与聚合物药物偶联物(polymerdrugconjugates)相比,小分子两亲性前药可以比较容易地合成,具有特定的化学结构,和高的药物负载能力。然而,很多小分子两亲性前药利用喜树碱的内酯环上的羟基作为链接基团,并形成相当稳定的酯键,这导致极其缓慢的释放和显著减少对癌细胞的细胞毒性。此外,由于临界聚集浓度较高,药物两亲物可能不能在体内维持其纳米结构,进而不能受益于纳米尺寸载体的增强的渗透性和保留(epr)效应。
白蛋白是血液中最常见的蛋白质,约7纳米大小,具有约20天的长血液半衰期。由于比较大的尺寸,纳米结构的epr效应的最早就是通过使用白蛋白和伊文思蓝(evansblue)结合发现的。因此,我们发明了一种能够利用白蛋白做蛋白载体并在细胞内迅速释放喜树碱两亲性小分子前药。在本发明中,喜树碱两亲性小分子前药在体内体外均能维持纳米结构,并能够在细胞内迅速释放。目前,国内外尚无这种两亲性小分子前药被报道。
技术实现要素:
本发明的首要目的是提供一种喜树碱前药,可作为小分子两亲性药物,在体内外均具有优异的肿瘤细胞摄取,并体现出显著的癌细胞抑制效果。
本发明的另一目的是提供制备所述的喜树碱前药的方法。
本发明的再一目的是提供所述的喜树碱前药在制备癌症治疗药物中的应用。
本发明的上述目的通过以下技术方案实现:
首先,提供一种喜树碱类前药,它是喜树碱或其衍生物和伊文思蓝形成的前药,其结构式如式(i)
其中,
r1为喜树碱或其衍生物基团;
r2为-ch2-,或者-o-中的一种;
r3为-ch2-,
x为s或者-ch2-中的一种;
n1,n2为重复单元数,均为0-10的整数。
本发明优选的方案中,所述的喜树碱或其衍生物基团来自式(ii)或式(iii)所示结构:
式(ii)为喜树碱的化学结构式,式(iii)为7-乙基-10-羟基喜树碱化学结构式。
本发明优选的方案中,所述的式(i)中的n1和n2均为0-5的整数;进一步优选0-2的整数。
本发明进一步优选的方案中,所述的式(i)中r1为喜树碱基团,r2为-o-或-ch2-,r3为
本发明更优选的方案中,所述的喜树碱类前药是结构如下式(iv)、(v)、(vi)或(vii)所示的任意一种:
本发明还提供一种制备所述的喜树碱类前药的方法,是以喜树碱或其衍生物为原料,在有机溶剂中,通过与带有相应基团的原料反应,制成喜树碱或其衍生物前药中间体,最后所述中间体再和伊文思蓝反应制备得到所述的喜树碱类前药;所述的喜树碱或其衍生物优选自喜树碱或7-乙基-10-羟基喜树碱。
本发明所述的制备方法中,制备所述的式(iv)或(v)的喜树碱类前药的方法,记作制备方法i,具体包括以下步骤:
ia)在有机溶剂中,在酰化催化剂存在下使用三光气活化喜树碱内酯环上的羟基,再加入过量的2,2'-二硫代二乙醇或1,6-己二醇反应,得到中间体醇;
ib)在催化剂存在下使用琥珀酸酐将步骤ia)得到的中间体醇转化为羧酸封端的中间体酸;
ic)将步骤ib)得到的中间体酸和伊文思蓝eb-nh2在有机溶剂中混合,并加入缩合剂搅拌反应,得到本发明所述的部分喜树碱类前药。
本发明所述制备方法i中,步骤ia)所述的有机溶剂可以选自二氯甲烷、氯仿、四氢呋喃、1,4-二氧六环或二甲基甲酰胺中的任意一种;优选二氯甲烷。
本发明所述制备方法i中,步骤ia)所述的酰化催化剂可以选自4-(二甲基氨基)吡啶、三乙胺或n,n-二异丙基乙胺中的任意一种;优选4-(二甲基氨基)吡啶。
本发明所述制备方法i中,步骤ib)所述的催化剂可以选自4-(二甲基氨基)吡啶、三乙胺或n,n-二异丙基乙胺中的任意一种;优选4-(二甲基氨基)吡啶。
本发明所述制备方法i中,步骤ic)所述的有机溶剂可以选自二甲基甲酰胺或二甲基亚砜中的任意一种;优选二甲基甲酰胺。
本发明所述制备方法i中,步骤ic)所述的缩合剂可以选自1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐(pybop)、n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)脲六氟磷酸盐(hbtu)或2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)中的任意一种或两种的混合物;优选1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐。
本发明所述制备方法i优选的一种实施方式中,制备了式(iv)所示的前药,其合成路线如下:
具体包括如下步骤:
在4-(二甲基氨基)吡啶的存在下,在二氯甲烷中,使用三光气活化喜树碱的羟基,然后与过量的2,2'-二硫代二乙醇反应,制备得到cpt-ss-oh;然后使用琥珀酸酐在有机溶剂中将cpt-ss-oh转化为羧酸封端的cpt-ss-cooh,其中需要添加4-(二甲基氨基)吡啶作为催化剂;最后,将cpt-ss-cooh和伊文思蓝eb-nh2一起在二甲基甲酰胺中混合,并加入1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐(pybop)和n,n-二异丙基乙胺(dipea)一起搅拌,制备得到式(iv)所示前药:cpt-ss-eb。
本发明所述制备方法i优选的另一种实施方式中,制备了以下式(v)所示的前药:
其合成路线如下:
具体包括如下步骤:
在4-(二甲基氨基)吡啶的存在下,在二氯甲烷中,使用三光气活化喜树碱的羟基,然后与过量的1,6-己二醇反应,制备得到cpt-cc-oh;然后使用琥珀酸酐在有机溶剂中将cpt-cc-oh转化为羧酸封端的cpt-cc-cooh,其中需要添加4-(二甲基氨基)吡啶作为催化剂;最后,将cpt-cc-cooh和伊文思蓝eb-nh2一起在二甲基甲酰胺中混合,并加入1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐(pybop)和n,n-二异丙基乙胺(dipea)一起搅拌,制备得到式(v)所示前药:cpt-cc-eb。
本发明所述的制备方法中,制备所述的式(vi)或(vii)的喜树碱类前药的方法,记作制备方法ii,具体包括以下步骤:
iia)在有机溶剂中,在酰化催化剂存在下,体系中加入缩合剂使喜树碱内酯环上的羟基与过量的3,3'-二氢氧啉酸或辛二酸一端的羧基发生缩合反应,得到羧基封端的中间体酸;
iib)将步骤iia)得到的中间体酸和伊文思蓝eb-nh2在有机溶剂中混合,并加入缩合剂搅拌反应,得到本发明所述的部分喜树碱类前药。
本发明所述制备方法ii中,步骤iia)所述的有机溶剂可以选自二氯甲烷、氯仿、四氢呋喃、1,4-二氧六环或二甲基甲酰胺中的任意一种;优选四氢呋喃或二氯甲烷。
本发明所述制备方法ii中,步骤iia)所述的酰化催化剂可以选自4-(二甲基氨基)吡啶、三乙胺或n,n-二异丙基乙胺中的任意一种;优选4-(二甲基氨基)吡啶。
本发明所述制备方法ii中,步骤iia)所述的缩合剂优选1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐。
本发明所述制备方法ii中,步骤iib)所述的有机溶剂可以选自二甲基甲酰胺或二甲基亚砜中的任意一种;优选二甲基甲酰胺;
本发明所述制备方法ii中,步骤iib)所述的缩合剂可以选自1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐(pybop)、n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)脲六氟磷酸盐(hbtu)或2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)中的任意一种或两种的混合物;优选1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐。
本发明所述制备方法ii优选的一种实施方式中,制备了以下式(vi)所示的前药:
其合成路线如下:
具体包括如下步骤:
在4-(二甲基氨基)吡啶的存在下,在二氯甲烷中,使用1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edchcl)作为缩合剂,然后与过量的3,3'-二氢氧啉酸反应,制备得到cpt-ss-cooh-2;最后,将cpt-ss-cooh-2和伊文思蓝eb-nh2一起在二甲基甲酰胺中混合,并加入1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐(pybop)和n,n-二异丙基乙胺(dipea)一起搅拌,制备得到式(vi)所示前药:cpt-ss-eb-2。
本发明所述制备方法ii优选的另一种实施方式中,制备了以下式(vii)所示的前药:
其合成路线如下:
具体包括如下步骤:
在4-(二甲基氨基)吡啶的存在下,在二氯甲烷中,使用1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edchcl)作为缩合剂,然后与过量的辛二酸反应,制备得到cpt-cc-cooh-2;最后,将cpt-cc-cooh-2和伊文思蓝eb-nh2一起在二甲基甲酰胺中混合,并加入1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐(pybop)和n,n-二异丙基乙胺(dipea)一起搅拌,制备得到式(vii)所示前药:cpt-cc-eb-2。
本发明还提供所述的喜树碱类前药在制备癌症治疗药物中的应用。
本发明设计和合成的喜树碱类前药是新型药物两亲性前药,在体外能够自组装成纳米粒子,在体内和白蛋白结合,形成两亲性前药/白蛋白复合体。本发明所述的喜树碱类药物两亲性前药制备简单,具有确定的化学结构和固定的高药物量,而且可以直接重悬浮于水溶液中,并自组装成直径为80±16nm的明确界定的纳米颗粒。纳米颗粒的形成提供了具有高水分散性的前药,并且保护药物免于水解。因此本发明所述的喜树碱类药物两亲性前药在水溶液中稳定,6天内几乎没有变化。试验证明本发明的喜树碱类两亲性前药能被癌细胞有效内吞,并且cpt-ss-eb、cpt-ss-eb-2等二硫键连接的前药能够在多种类型的癌细胞中的体现很强的细胞毒性,ic50值比伊立替康低10-27倍。此外cpt-ss-eb、cpt-ss-eb-2等的二硫键能够在谷胱甘肽存在下迅速裂解并释放出喜树碱,从而能够维持其抗癌活性。
现有技术中,与聚合物-药物的形成纳米颗粒相比,小分子药物两亲性前药的一个潜在缺点是其在体内血液循环中在稀释条件下的相对低的稳定性。但是本发明中,通过eb-白蛋白结合,cpt-ss-eb两亲性前药能够从80nm纳米颗粒瞬时转化为7nm的白蛋白/前药复合物。体内pet成像研究证实,本发明中的药物两亲性前药具有很长的血液循环,并能够在肿瘤中富集。在注射后24小时,cpt-eb的半衰期和曲线下面积(auc)比喜树碱改善了130倍和30倍。cpt-ss-eb的肿瘤积累相对于喜树碱增加了40倍。同时,本发明的小分子两亲性前药对结肠癌具有优异的抗癌功效。总体而言,本发明制备的小分子两亲性前药具有显著的创新性和极强的临床转化能力,为小分子药物递送系统的开发开辟了一种新的途径。
附图说明
图1为cpt-ss-oh的1hnmr(300mhz,cd2cl2)。
图2为cpt-ss-cooh的1hnmr(300mhz,cdcl3)。
图3为cpt-ss-eb的1hnmr(300mhz,dmso-d6)。
图4为cpt-ss-eb的hplc和esi-ms谱图。
图5为cpt-cc-oh的1hnmr(300mhz,cdcl3)。
图6为cpt-cc-cooh的1hnmr(300mhz,cdcl3)。
图7为cpt-cc-eb的hplc和esi-ms谱图。
图8为cpt-nh2的1hnmr(300mhz,cd2cl2)。
图9为eb-nota-cpt的hpl谱图。
图10为eb-nota-cpt的lc-mass谱图。
图11为nota-cpt的lc-mass谱图。
图12为cpt-ss-eb临界聚集浓度的测定。
图13为cpt-ss-eb的流体动力学直径变化图。
图14为cpt-ss-eb和cpt-cc-eb的在37度ph7.4条件下搅拌3天后的hplc谱图。
图15为cpt-ss-eb加入白蛋白以后的伊文思荧光变化图。
图16为cpt-eb和cpt的血液半衰期。
图17为cpt-ss-eb前药的表征。(a)cpt-ss-eb在水中的照片(左)和红色激光通过后(右)的照片;(b)cpt-ss-eb在pbs中的水合直径分布图;(c)cpt-ss-eb的tem图像;(e)具有或不具有白蛋白的cpt-ss-eb和eb-cooh的荧光光谱;(d)加入白蛋白5分钟后cpt-ss-eb的水合直径分布图和(f)加入白蛋白24小时后的cpt-ss-eb的水合直径分布图。
图18为37℃下在pbs中,在含有/不含10mm谷胱甘肽(gsh)的pbs中的喜树碱(cpt)的释放。
图19为cpt-ss-eb、cpt-cc-eb、ir和cpt不同的癌细胞(a)hct116,(b)u87mg,(c)4t1,和(d)a549的体外细胞毒性。
图20为共焦显微镜图像显示(a)cpt-cc-eb和(b)cpt-ss-eb的细胞内吞。蓝色=hoechst(核染色),绿色=lysogreen跟踪剂(溶酶体),红色=来自前药的eb。
图21为cpt-ss-eb和相关化合物的流式细胞仪分析结果。左:450nm通道,对应于来自cpt的荧光;右:660nm通道,对应于来自eb的荧光。
图22为静脉注射[64cu]标记的(a)cpt-eb和(b)cpt后5分钟,1小时,3小时,5小时,7小时和24小时的hct116肿瘤携带小鼠的代表性全身冠状和横向pet图像。在示踪剂注射后不同时间点的cpt-eb和cpt在心脏(c,具有血液),肿瘤(d)和肝脏(e)中的分布。定量基于pet图像(n=3/组)。(f)在注射后48小时杀死小鼠,收集他们的器官和组织的定量生物分布数据。白色箭头/圆圈表示肿瘤的位置。数据表示为平均值±标准误差(n=3/组)。
图23为hct116肿瘤生长抑制图,每组5只小鼠,每3天一次,注射5次。
具体实施方式
下面通过实施例对本发明进行具体描述,本实施例只用于对本发明作进一步的说明,不能理解为对本发明保护范围的限制,本领域的技术人员根据上述发明的内容做出的一些非本质的改进和调整,均属本发明保护范围。
实施例1:cpt-ss-oh的制备
在搅拌下,将4-二甲氨基吡啶(1.05g,8.60mmol)在10ml二氯甲烷中的溶液滴加到喜树碱(1.0g,2.87mmol)和三光气(0.315g,1.06mmol)的无水二氯甲烷(200ml)的混合物悬浮液中。搅拌30分钟后,加入在无水四氢呋喃(25ml)中的2,2'-二硫代二乙醇(8.60g,55.8mmol),将反应混合物在室温下搅拌过夜。将混合物用50mm盐酸水溶液(2×100ml),水(1×100ml)和饱和盐水(1×100ml)洗涤。分离有机层并用无水硫酸钠干燥。通过旋转蒸发器浓缩溶液,并使用预填充的二氧化硅柱通过快速色谱分离纯化。产量:1.05g(69%产率)。1hnmr(300mhz,cd2cl2)δ8.46(s,1h),8.22(d,j=8.3hz,1h),7.99(dd,j=8.2,1.1hz,1h),7.86(ddd,j=6.9,6.5hz,1h),7.70(ddd,j=8.1,6.9,1.2hz,1h),7.42(s,1h),5.64(d,j=18hz,1h),5.36(m,1h)4.37(t,j=6.0hz,2h),3.93-3.81(m,2h),3.03-2.82(m,4h),2.19(tdd,j=14.0,12.3,7.5hz,2h),1.01(t,j=7.5hz,3h)。图1为cpt-ss-oh的1hnmr谱图,证明了制备该化合物成功。
实施例2:cpt-ss-cooh的制备
在搅拌下将琥珀酸酐(400mg,4.00mmol)、实施例1制备的cpt-ss-oh(204mg,0.386mmol)和4-二甲氨基吡啶(19.6mg,0.161mmol)溶于无水二氯甲烷(200ml)中。将反应混合物在室温下搅拌过夜,然后用水(1×100ml),50mm盐酸水溶液(1×100ml)和饱和盐水(1×100ml)洗涤。分离有机层并用无水硫酸钠干燥。通过旋转蒸发器浓缩溶液,并使用预填充的二氧化硅柱通过快速色谱纯化。产量:201mg(83%产率)。1hnmr(300mhz,cdcl3)δ8.44(s,1h),8.32(d,j=8.4hz,1h),7.95(d,j=8.2hz,1h)(ddd,j=8.5,6.9,1.4hz,1h),7.69(ddd,j=8.1,7.0,1.1hz,1h),7.43(s,1h),5.71(d,j=17.3hz,1h)5.39(d,j=17.3hz,1h),5.32(s,2h),4.46-4.25(m,4h),3.00-2.86(m,4h),2.79-2.61m,2h),2.18(m,j=2h),1.01(t,j=7.5,3h)。esi-msm/z:计算值628,实测值629(m+h)。图2为cpt-ss-cooh的1hnmr谱图,证明了制备该化合物成功。
实施例3:cpt-ss-eb的制备
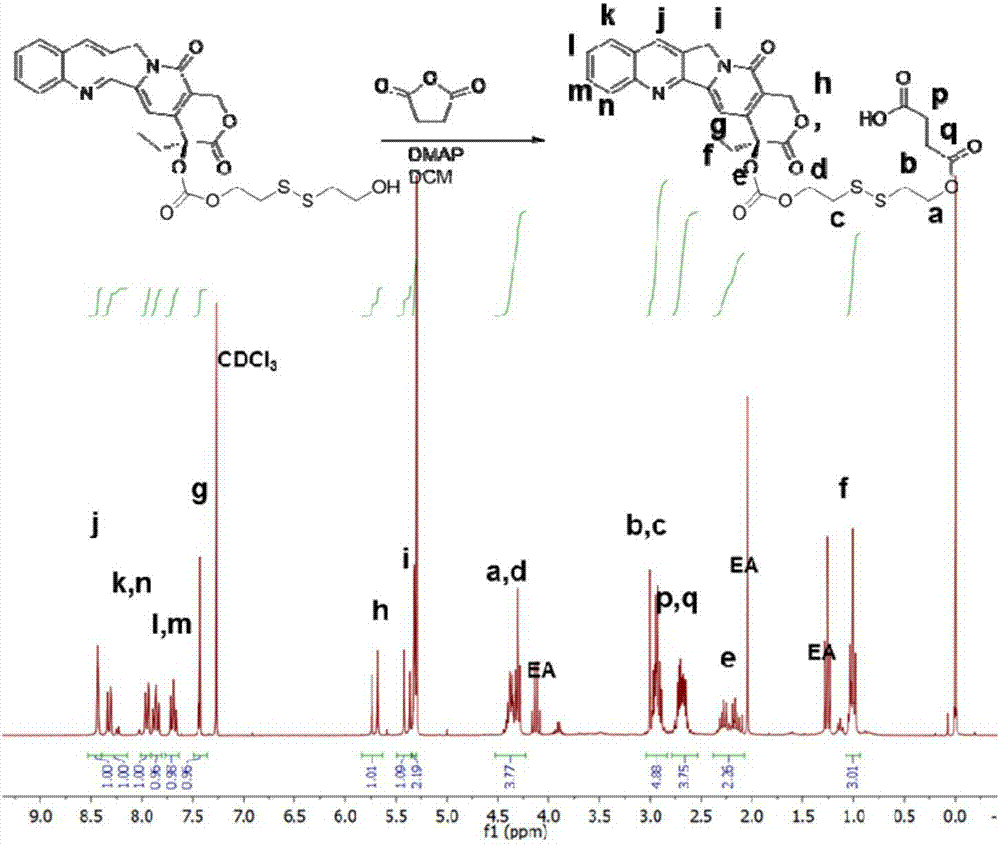
实施例2制备的cpt-ss-cooh(58mg,0.092mmol)、eb-胺(25mg,0.046mmol)、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基鏻(pybop,48mg,0.092mmol)和n,n-二异丙基乙胺(dipea,59mg,0.46mmol)在二甲基甲酰胺中混合,在氮气下搅拌2天。通过加入过量的乙酸淬灭反应,并通过制备型高效液相使用乙腈和0.2%乙酸水溶液(梯度:5-95%乙腈)纯化。将收集的纯化产物冻干并储存在-20℃以备后用。产量:21mg(40%产率)。esi-msm/z:计算值1152,实测值1151(m-h)。1hnmr(300mhz,dmso)δ9.34(s,1h),8.70(d,j=3.7hz,1h),8.35(s,1h),8.21-8.10(m,3h),8.02(d,9.9hz,1h),7.88(dt,j=8.4,4.1hz,2h),7.73(dd,j=6.9,4.1hz,1h),7.64(dd,j=4.0hz,j=6.5hz,1h),7.50(d,j=5.2hz,2h),7.09(d,j=2.2hz,1h),7.00(d,j=10hz,1h),5.56-5.32(m,4h),4.32(t,j=5.9hz,2h),4.21(t,j=6.2hz,2h),3.55(t,j=6.4hz,1h),3.06-2.89(m,3h),2.75(dd,j=11.9,5.4hz,1h),2.66-2.52(m,6h),2.28(s,3h),2.26(s,3h),2.22-2.13(m,4h),0.92(t,j=7.4hz)。图3为cpt-ss-eb的1hnmr谱图,证明了制备该化合物成功。图4为cpt-ss-eb的hplc和esi-ms谱图,也证明了该化合物成功制备,并且具有99%以上纯度。
实施例4:cpt-cc-oh的制备
在搅拌下,将4-二甲氨基吡啶(1.68g,13.8mmol)在10ml二氯甲烷中的溶液滴加到喜树碱(1.5g,4.31mmol)和三光气(0.473g,1.59mmol)的无水二氯甲烷(200ml)的混合物悬浮液中。搅拌30分钟后,加入在无水四氢呋喃(15ml)中的1,6-己二醇(6.64g,43.1mmol),将反应混合物在室温下搅拌过夜。将混合物用50mm盐酸水溶液(2×100ml),水(1×100ml)和饱和盐水(1×100ml)洗涤。分离有机层并用无水硫酸钠干燥。通过旋转蒸发器浓缩溶液,并使用预填充的二氧化硅柱通过快速色谱纯化。使用梯度乙酸乙酯和己烷混合物作为洗脱剂。产量:1.48g(70%产率)。1hnmr(300mhz,cdcl3)δ8.41(s,1h),8.23(d,j=8.7hz,1h),7.95(dd,j=8.2,1.1hz,1h),7.85(ddd,j=6.9,6.5hz,1h),7.68(ddd,j=8.1,6.9,1.2hz,1h),7.36(s,1h),5.70(d,j=17.3hz,1h),5.39(d,j=1h),5.30(s,2h),4.21-4.05(m,2h),3.59(dd,j=10.1,6.1hz,2h),2.43-2.01(m,2h),1.74-1.65(m,3h),1.59-1.47(m,2h),1.45-1.32(m,4h),1.00(t,j=7.5hz,3h)。图5为cpt-cc-oh的1hnmr谱图,证明了制备该化合物成功。
实施例5:cpt-cc-cooh的制备
在搅拌下将琥珀酸酐(400mg,4.00mmol)、实施例4制备的cpt-cc-oh(200mg,0.406mmol)和4-二甲氨基吡啶(19.8mg,0.162mmol)溶于无水二氯甲烷(200ml)中。将反应混合物在室温下搅拌过夜,然后用水(1×100ml),50mm盐酸水溶液(1×100ml)和饱和盐水(1×100ml)洗涤。分离有机层并用无水硫酸钠干燥。通过旋转蒸发器浓缩溶液,并使用预填充的二氧化硅柱通过快速色谱纯化。使用梯度乙酸乙酯和己烷混合物作为洗脱剂。产量:192mg(80%产率)。esi-msm/z(m+)计算值592,实测值591(m-h)。1hnmr(300mhz,cdcl3)δ8.42(s,1h),8.28(d,j=8.7hz,1h),7.95(d,j=8.3hz,1h),7.85(ddd,j=1h),7.68(ddd,j=8.1,6.9,1.1hz,1h),7.38(d,j=5.1hz,1h),5.70(d,j=17.3hz,1h),5.39(d,j=17.3hz,1h),5.31(s,2h),4.23-3.99(m,4h),2.74-2.56(m,4h),2.37-2.07(m,2h),1.61(ddd,j=12.9,6.6hz,5h),1.44-1.23(m,7h),1.00(t,j=7.5hz,3h)。图6为cpt-cc-cooh的1hnmr谱图,证明了制备该化合物成功。
实施例6:cpt-cc-eb的制备
将实施例5制备的cpt-cc-cooh(52mg,0.092mmol)、eb-胺(25mg,0.046mmol)、六氟磷酸苯并三唑-1-基-氧三吡咯烷基鏻(pybop,48mg,0.092mmol)和n,n-二异丙基乙胺(dipea,59mg,0.46mmol)在二甲基甲酰胺中混合,在氮气下搅拌2天。通过加入过量的乙酸淬灭反应,并通过制备型高效液相使用乙腈和0.2%乙酸水溶液(梯度:5-95%乙腈)纯化。将收集的纯化产物冻干并储存在-20℃以备后用。产量:23mg(45%产率)。esi-msm/z:计算值1116,实测值1115(m-h)。图7为cpt-cc-eb的hplc和esi-ms谱图,证明了该化合物成功制备,并且具有99%以上纯度。
实施例7:cpt-ss-cooh-2的制备
在搅拌下将3,3'-二氢氧啉酸(1197mg,5.7mmol)、cpt(200mg,0.57mmol)和4-二甲氨基吡啶(35mg,0.29mmol)混于四氢呋喃(200ml)中。将反应混合物在室温下搅拌过夜,然后用饱和碳素氢钠水溶液(3×100ml),50mm盐酸水溶液(1×100ml)和饱和盐水(1×100ml)洗涤。分离有机层并用无水硫酸钠干燥。通过旋转蒸发器浓缩溶液,并使用预填充的二氧化硅柱通过快速色谱纯化。使用梯度乙酸乙酯和己烷混合物作为洗脱剂。产量:121mg(39%产率)。1hnmr(300mhz,cdcl3)δ8.41(s,1h),8.27(d,j=8.7hz,1h),7.95(d,j=8.3hz,1h),7.85(m,1h),7.68(m,1h),7.37(d,j=5.1hz,1h),5.70(d,j=17.2hz,1h),5.39(d,j=17.2hz,1h),5.30(s,2h),2.92-2.59(m,8h),2.36-2.06(m,2h),1.01(t,j=7.5hz,3h)。
实施例8:cpt-ss-eb-2的制备
实施例7制备的cpt-ss-cooh-2(50mg,0.092mmol)、eb-胺(25mg,0.046mmol)、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基鏻(pybop,48mg,0.092mmol)和n,n-二异丙基乙胺(dipea,59mg,0.46mmol)在二甲基甲酰胺中混合,在氮气下搅拌2天。通过加入过量的乙酸淬灭反应,并通过制备型高效液相使用乙腈和0.2%乙酸水溶液(梯度:5-95%乙腈)纯化。将收集的纯化产物冻干并储存在-20℃以备后用。产量:11mg(22%产率)。esi-msm/z:计算值1064,实测值1063(m-h)。1hnmr(300mhz,dmso)δ9.35(s,1h),8.71(d,j=3.7hz,1h),8.35(s,1h),8.21-8.11(m,3h),8.03(d,9.9hz,1h),7.89(m,2h),7.74(m,1h),7.65(m,1h),7.51(d,j=5.2hz,2h),7.09(d,j=2.3hz,1h),7.01(d,j=10.0hz,1h),5.56-5.31(m,4h),2.98-2.52(m,8h),2.28(s,3h),2.25(s,3h),2.22-2.13(m,2h),0.91(t,j=7.4hz)。
实施例9:cpt-cc-cooh-2的制备
在搅拌下将辛二酸(991mg,5.7mmol)、cpt(200mg,0.57mmol)和4-二甲氨基吡啶(35mg,0.29mmol)混于四氢呋喃(200ml)中。将反应混合物在室温下搅拌过夜,然后用饱和碳素氢钠水溶液(3×100ml),50mm盐酸水溶液(1×100ml)和饱和盐水(1×100ml)洗涤。分离有机层并用无水硫酸钠干燥。通过旋转蒸发器浓缩溶液,并使用预填充的二氧化硅柱通过快速色谱纯化。使用梯度乙酸乙酯和己烷混合物作为洗脱剂。产量:109mg(38%产率)。1hnmr(300mhz,cdcl3)δ8.42(s,1h),8.27(d,j=8.7hz,1h),7.96(d,j=8.3hz,1h),7.86(m,1h),7.68(m,1h),7.38(d,j=5.1hz,1h),5.71(d,j=17.3hz,1h),5.38(d,j=17.3hz,1h),5.31(s,2h),2.46-2.08(m,6h),1.68-1.21(m,8h),1.00(t,j=7.5hz,3h)。
实施例10:cpt-cc-eb-2的制备
实施例9制备的cpt-cc-cooh-2(46mg,0.092mmol)、eb-胺(25mg,0.046mmol)、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基鏻(pybop,48mg,0.092mmol)和n,n-二异丙基乙胺(dipea,59mg,0.46mmol)在二甲基甲酰胺中混合,在氮气下搅拌2天。通过加入过量的乙酸淬灭反应,并通过制备型高效液相使用乙腈和0.2%乙酸水溶液(梯度:5-95%乙腈)纯化。将收集的纯化产物冻干并储存在-20℃以备后用。产量:12mg(21%产率)。esi-msm/z:计算值1029,实测值1028(m-h)。1hnmr(300mhz,dmso)δ9.33(s,1h),8.70(d,j=3.7hz,1h),8.34(s,1h),8.20-8.10(m,3h),8.01(d,9.9hz,1h),7.87(m,2h),7.72(m,1h),7.63(m,1h),7.50(d,j=5.2hz,2h),7.08(d,j=2.2hz,1h),7.00(d,j=10hz,1h),5.56-5.31(m,4h),2.49-2.02(m,12h),1.68-1.21(m,8h),0.92(t,j=7.4hz)。
实施例11:cpt-nhs活化酯的制备
在-10℃下将实施例5制备的cpt-cc-cooh(27μmol)和n-羟基琥珀酰亚胺(27μmol)溶解在1.0ml无水thf中。然后滴加n,n'-二环己基碳二亚胺(dcc,27μmol)在无水四氢呋喃(0.5ml)中的溶液。将反应混合物在-10℃下搅拌1小时,然后在室温下搅拌过夜。通过过滤除去白色沉淀,真空除去溶剂,得到油状物。将产物在乙酸乙酯中重结晶,得到白色-黄色固体产物(52%产率)。
实施例12:eb-nota-cpt的制备
将eb-lys-nota(5.0μmol)溶解在200μl二甲基甲酰胺中。然后加入在100μl二甲基甲酰胺中的cpt-nhs活化酯(实施例11制备的)(6.0μmol),然后加入n,n-二异丙基乙胺(dipea,25μmol)。将反应在室温下搅拌3-4小时。然后,使用higgins柱纯化粗产物。获得化学纯度>95%的产物。lcms1529.46(m-h)。图9,图10为eb-nota-cpt的hplc和lc-mass谱图,证明了该化合物成功制备,并且具有95%以上纯度。
实施例13:cpt-nh2的制备
向在1ml无水二甲基甲酰胺(dmf)中的实施例5制备的cpt-cc-cooh(3.6当量)溶液中加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯((hatu,4.2当量)。将溶液在室温下搅拌10分钟。然后加入10当量的n,n-二异丙基乙胺,然后加入n-叔丁氧羰基-1,2-乙二胺(1.0当量)的二甲基甲酰胺溶液。将反应在室温下搅拌过夜。使用油真空泵除去二甲基甲酰胺,并使用二氯甲烷/水萃取粗混合物。蒸发有机层,然后在室温下用三氟乙酸/二氯甲烷(1:1)处理1小时。将混合物在高效液相制备柱上纯化,得到cpt-nh2,为黄色固体(44%产率)。图8为cpt-nh2的1hnmr谱图,证明了制备该化合物成功。
实施例14:nota-cpt的制备
将cpt-nh2(5.0μmol)溶解在200μl二甲基甲酰胺中。然后加入在100μl二甲基甲酰胺中的nota-nhs活化酯(6.0μmol),然后加入n,n-二异丙基乙胺(dipea,25μmol)。将反应在室温下搅拌3-4小时。然后,使用higgins柱方法纯化。产物以>95%的化学纯度。lcms:920.34(m+h)使用分析型高效液相和lc-ms来确认产物已经纯化。图11为nota-cpt的hplc和lc-mass谱图,证明了该化合物成功制备,并且具有95%以上纯度。
实施例15:临界聚集浓度(cac)测量
芘被用作为荧光探针,将实施例3制备的cpt-ss-eb以1.0mg/ml分散在水中,芘含量为6.0×10-7mol/l。然后将溶液以6.0×10-7mol/l的恒定芘浓度稀释至284μg/ml至0.277μg/ml的各种浓度。记录所有样品的激发光谱,发射波长设定在375nm。测定所有溶液的i339/i335比率值,并且使用这些值相对于对数刻度(logc)上的浓度作图。在cpt-ss-eb浓度<1.5μg/ml时,强度比为约0.25,表明芘在亲水环境中。当浓度>1.5μg/ml时,强度比显着增加,最终达到~0.9,这是疏水环境中芘的特征。图12为测得结果,显示临界聚集浓度值在水为1.5μg/ml。
实施例16:cpt-ss-eb在水中的组装形成纳米粒子和相关表征
冻干的cpt-ss-eb可以直接重悬在水或其他水溶液中,并且由于其内在的两亲性质而自发地自组装成纳米颗粒。由于eb的优异的水溶性和纳米颗粒的形成,cpt-ss-eb可以以很高的浓度(5mg/ml)分散在水溶液中(参见图17a左)。当红色激光通过cpt-ss-eb溶液时,显示强的光散射,这表明纳米颗粒已经形成(参见图17a右)。我们进一步用动态光散射测定了cpt-ss-eb水合直径。结果显示纳米颗粒具有80±16nm的数均平均流体动力学直径(图17b)。透射电子显微镜(tem)表明cpt-ss-eb纳米颗粒是球形的,具有约57nm的相对均匀的尺寸(图17c)。稳定的纳米颗粒的形成不仅增强了喜树碱的水分散性,而且保护了其不被水解。如由dls在6天内监测,纳米组装件在pbs中保持流体动力学二聚体,进一步指示其稳定性(图13)。此外,hplc也用于证实本发明的药物两亲物的稳定性。在pbs中孵育三天后,仅观察到痕量的降解产物,并且对于实施例3制备的cpt-ss-eb和实施例6制备的cpt-cc-eb这两种药物两亲物均没有检测到游离cpt(参见图18中的方块和倒三角标记的两条曲线)。在水性环境中的高稳定性,连同优异的重复冻干和再悬浮的能力使得本发明的药物两亲物作为药物递送平台具有高度吸引力。
实施例17:cpt-ss-eb在水中的组装形成纳米粒子和白蛋白的相互作用
现有技术中,虽然有些小分子药物两亲性前药在水溶液中可能是稳定的,但它们可能不能在体内保持其完整性。本发明的cpt-ss-eb两亲性前药的一个关键优点是通过伊文斯蓝与白蛋白结合,将它们转化为白蛋白与药物的复合物。新形成的白蛋白/药物复合物仍然是纳米结构,并利用白蛋白的长血液循环和epr效应优先将药物积累在肿瘤。一系列研究验证了cpt-ss-eb与白蛋白结合的能力。首先,我们发现在过量牛血清白蛋白(bsa)存在下cpt-ss-eb的eb荧光强度增加了10倍,程度与未偶联的eb-cooh相当(参见图17e中上部的两条曲线)。
我们进一步进行了白蛋白结合的荧光动力学研究,将0.2mg/ml(对应于药物注射后的初始小鼠血浆药物浓度)的cpt-ss-eb的eb荧光监控20分钟,然后加入白蛋白,终浓度为35mg/ml(对应于小鼠血清白蛋白浓度)。加入白蛋白后,eb荧光增加7.3倍,并在4小时内逐渐增加总共8.6倍,变化趋势参见图15。
通过动态光散射测量,在加入白蛋白后,cpt-ss-eb纳米组装体的数均平均流体动力学直径从80nm变为7nm,表明大多数cpt-ss-eb从较大纳米结构解离并转化为白蛋白/cpt-ss-eb复合物。此外,对大纳米颗粒非常敏感的强度平均流体动力学直径从用于cpt-ss-eb纳米组装的121nm变化为加入白蛋白之后的148nm和7nm,表明同时存在白蛋白/cpt-ss-eb复合物以及一小部分剩余的cpt-ss-eb纳米组装。1天后,数量和强度平均的流体动力学直径均变为约7nm。这些结果表明,cpt-ss-eb纳米组件通过白蛋白结合cpt-ss-eb而固定地转化为白蛋白/cpt-ss-eb复合物。
实施例18:喜树碱释放实验
通过透析法测量喜树碱从cpt-cc-eb和cpt-ss-eb的释放。简言之,将水中的cpt-cc-eb或cpt-ss-eb用磷酸盐缓冲盐水(pbs)稀释,产生55μg/ml的浓度,将0.5ml转移到预浸渍的透析盒中,放置于25ml磷酸盐缓冲盐水或者含有10mm谷胱甘肽的磷酸盐缓冲盐水中透析72小时。在预定时间从透析液中取等分试样(0.1ml),并将0.1ml新鲜磷酸盐缓冲盐水加回到外部透析液中。通过高效液相(条件:30%乙腈,含有0.1%tfa,1ml/min,uv检测器,250nm)监测喜树碱浓度的增加。
结果发现(如图18所示),cpt-ss-eb在不含gsh的pbs中非常稳定,并且在72小时内仅释放~3%的cpt(图18中接近横坐标的方块标记的曲线)。相比之下,在gsh的存在下,超过80%的喜树碱在12小时内快速释放(图18中最高处的原点标记的曲线)。该结果表明二硫键容易被gsh切割,其随后导致碳酸酯键的分解以产生游离的喜树碱。这些结果同时表明,cpt-ss-eb在正常生理条件(例如盐水,血液和细胞外基质)中相对稳定,但在细胞内,特别是具有升高的gsh水平的癌细胞中快速重新释放高毒性cpt。如我们所预期的,没有二硫键,cpt-cc-eb在pbs或单独的10mmgsh中均释放有限量(<5%)的cpt。
实施例19:体外细胞毒性测定
将人神经胶质瘤细胞系u87mg,人结肠癌细胞hct116,人肺癌细胞系a549或小鼠乳腺癌4t1接种在96孔板中。将细胞在37℃下在含有5%二氧化碳的潮湿气氛中孵育。接种后24小时用新鲜培养基替换培养基。将喜树碱等制剂溶解在溶剂中,并使用细胞培养基稀释。对于每个孔,加入100μl具有不同指定药物浓度的细胞培养基。将细胞孵育48小时,并且在此期间后,用含有0.5mg/ml的3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四氮唑(mtt)的100μl培养基替换培养基。细胞与试剂孵育2小时后,小心地除去含有未反应的mtt的培养基。然后,将获得的蓝色晶体溶解于100μldmso中,并在bioteksynergyh4混合读数器中在570nm的波长下测量吸光度。将空白减去测量的光密度(od)值,并且基于对照未处理细胞的相对吸光度计算细胞活力。使用graphpadprism5进行ic50值的计算和统计分析。
我们首先测试了对来自结肠癌的细胞系hct116的体外细胞毒性(图19a)。如我们所预期的,fda批准的ir显示出比cpt(0.087μm)高得多的ic50(1.8μm)。cpt-cc-eb显示出最低的细胞毒性,因为活性羟基转化成与eb的稳定酯键。cpt-cc-eb的低毒性还表明cpt-ss-eb在血液循环期间可能具有低毒性,其中gsh浓度低。cpt-ss-eb的ic50值确定为0.15μm,其类似于高效的cpt,但比cpt-cc-eb低近300倍。这表明细胞内gsh能够从还原敏感性药物两亲物cpt-ss-eb切割cpt。另外cpt-ss-eb-2的细胞毒性比cpt-ss-eb稍低(ic50为0.29μm),但也显著高于ir。cpt-cc-eb-2毒性与cpt-cc-eb类似。值得注意的是,u87mg对cpt-ss-eb和cpt也显示出非常高的灵敏度,ic50值分别为0.31μm和0.12μm(参见图19b)。我们还证实,cpt-ss-eb和cpt对4t1和a549癌细胞具有相似的细胞毒性(参见图19c、图19d),尽管与hct116细胞相比,所有制剂对这些细胞的敏感性相对较低。总之,这些研究表明本发明的喜树碱类前药不但都具有良好的水溶性和稳定性,而且其中的cpt-ss-eb、cpt-ss-eb-2等二硫键连接的前药与cpt具有相当的体外细胞毒性,并且比fda批准的ir更有效,并且可切割的连接体能够显著抑制癌细胞增殖。
实施例20:体外细胞摄取
使用共聚焦激光扫描显微镜和流式细胞仪研究喜树碱体外细胞摄取。将eb-ss-cpt和eb-cc-cpt(10μm)与hct116细胞孵育4小时,并用lysotrackergreen和10μg/mlhoechst33342在细胞培养箱中37℃孵育0.5小时,共聚焦观察。然后将细胞用磷酸盐缓冲盐水洗涤三次,之后在zeisslsm780共聚焦显微镜上成像。或者,对于流式细胞术分析,将hct116细胞接种到24孔板中,24小时后,用eb-ss-cpt、eb-cc-cpt、喜树碱、伊立替康和eb-胺(10μm每种药物)4小时。然后,使用胰蛋白酶分离细胞,并用磷酸盐缓冲盐水洗涤三次。使用bdbeckmancoulter流式细胞仪分析细胞的荧光强度。eb荧光:激发532nm,发射660nm;cpt荧光:激发-355nm,发射450nm。
由于eb是荧光的,特别是在与白蛋白结合后,这些药物两亲性前药可以直接成像,而不需要另外的染料标记。在hct116细胞的共聚焦显微镜中,细胞核通过hoechst染色,溶酶体使用lysotrackergreen染色。孵育4小时后,cpt-ss-eb和cpt-cc-eb定位于溶酶体和细胞核中(参见图20a-b,其中a体现cpt-cc-eb的细胞内吞;b体现cpt-ss-eb的细胞内吞),表明本发明的药物两亲物可通过内吞途径内化。我们进一步进行流式细胞术分析以尝试比较不同制剂的细胞摄取(图21)。根据喜树碱荧光,与游离喜树碱、伊立替康和未处理的对照相比,本发明的cpt-ss-eb和cpt-cc-eb两种药物两亲物具有高得多的细胞摄取(如图21中左图所示)。此外,基于eb荧光,本发明的cpt-ss-eb和cpt-cc-eb两种药物两亲性前药与eb和对照相比具有高细胞摄取(如图21中右图所示)。从上述实验结果可以得出,本发明的cpt-ss-eb等二硫键连接的前药和cpt-cc-eb等碳-碳键连接的前药,都可被hct116细胞有效内吞。
实施例21:hct116肿瘤的体内正电子发射计算机断层扫描(pet)成像。
通过皮下注射5×106个hct116细胞在磷酸盐缓冲盐水(100μl)中的悬浮液制备裸鼠hct116移植瘤:(7周龄,雌性)。当肿瘤大小达到500-1000mm3时,将小鼠用于pet成像。在示踪剂注射前使用异氟烷/氧气(2%v/v)麻醉小鼠。将麻醉的小鼠静脉内注射在磷酸盐缓冲盐水(100μl)中配置的64cu标记的实施例12制备的cpt-nota-eb和实施例14制备的cpt-nota(4.44-5.55mbq/120-150μci每只小鼠)。在注射后指定的时间点,在inveondpet扫描仪(siemensmedicalsolutions,malvern,pa)上扫描小鼠。使用3d有序子集期望最大化算法重建没有衰减或散射的校正的正电子发射计算机断层扫描图像。使用asiprovmtm软件进行图像分析。在任何感兴趣的器官上绘制感兴趣的区域(roi)以计算%id/g。
上述小鼠在注射后48小时处死。收集器官和血液并湿称重。将收集的器官和血液与一系列标准溶液一起在γ-计数器(wallacwizard1480,perkinelmer)上测量64cu放射性。将器官和血液的放射性转换以计算目标器官中的注射剂量(%id)的百分比和每克组织的注射剂量的百分比(%id/g)。
通过pet成像(图22),我们发现cpt-eb逐渐积累在肿瘤中(图22a),而cpt的没有明显积累(图22b)。我们进一步量化cpt-eb和cpt在心脏、肿瘤和肝脏的分布。如图22c所示,cpt-eb的浓度随时间逐渐降低,血液半衰期为~6.5小时(图16)。相比之下,cpt浓度迅速下降,血液半衰期仅为~3分钟。cpt-eb的曲线下面积(auc)为221.2±17.9h*%id/g,比cpt的面积(7.3±1.2h*%id/g)增大了30倍。半衰期的130倍增加和auc的30倍增加证明cpt-eb具有比cpt长得多的血液循环时间。更重要的是,cpt-eb的肿瘤摄取从5分钟时的1.18±0.24%id/g迅速增加到5小时的5.61±1.14%id/g,然后逐渐增加至6.33±0.22%id/24小时(如图22d所示)。对于cpt,观测到的则是相反的趋势:在注射后5分钟时最高浓度为0.79±0.27%id/g,并且在注射后24小时衰减至接近零(图22d)。cpt浓度的急剧下降可能是由于高的肝吸收,然后被消化并从身体排出(图22e)。通过γ-计数的体外定量的器官放射性还证实了注射后48小时的cpt-eb在肿瘤组织中的4.26±0.97%id/g比cpt在肿瘤组织中更有效的积累(图22f)。值得注意的是,在肾脏中有显著量的放射性,表明cpt-eb可能从可逆结合的白蛋白分离并通过肾清除从体内排出。这些体内药代动力学和生物分布清楚地表明本发明的药物两亲性前药具有较长的血液循环和较高的肿瘤富集。
实施例22:hct116荷瘤小鼠的体内治疗
通过皮下注射5×106个hct116细胞在磷酸盐缓冲盐水(100μl)中的悬浮液制备hct116荷瘤裸鼠(7周龄,雌性)。当在肿瘤接种后第10天建立肿瘤时,通过每3天静脉内注射100μl药物(cpt-ss-eb、cpt-cc-eb、ir、cpt)或磷酸盐缓冲盐水最多5次开始用相同方案治疗小鼠(按喜树碱的剂量计算:3mg/公斤)。每三天监测肿瘤体积和小鼠重量。当肿瘤的任何尺寸超过2cm或当小鼠体重减轻超过20%时,对小鼠实施安乐死。使用以下公式分别计算每种类型肿瘤的体积:体积=(长度×宽度2)/2。
如图23所示,cpt-ss-eb显示出最有效的抗肿瘤效果,能够显著延迟肿瘤发展(参见图23中接近横坐标的菱形标记的曲线)。cpt-cc-eb的抗肿瘤活性与fda批准的伊诺替康(ir)在治疗后22天内的抗肿瘤功效相当(参见图23中处于中间位置的菱形标记曲线和倒三角形标记曲线)。此外,很重要的一点是,用cpt-ss-eb和cpt-cc-eb治疗的小鼠在治疗过程中均生存良好,体重未明显下降;然而,用喜树碱(cpt)治疗的小鼠发生了严重减重(减少20%)和注射部位的损伤,并且由于副作用而必须在接种后第18天处死(参见图23中接近横坐标的三角形标记曲线)。这些结果进一步证明,本发明的喜树碱类前药的抗肿瘤效果达到甚至超过了现有的常规抗肿瘤药,尤其是具有二硫键的药物两亲性前药纳米组装体表现出了非常有效的治疗效果和低副作用。
- 还没有人留言评论。精彩留言会获得点赞!