一种无外源标记基因的抗PCV2转基因猪制备方法与流程
本发明涉及基因工程领域,特别涉及一种无外源标记基因的抗pcv2转基因猪制备方法。
背景技术:
在家畜动物的生长及疾病防治中,转基因技术日趋成熟,为便于转基因细胞系的筛选及转基因动物的检测,在转入目的基因的同时,常常同时转入外源标记基因,而egfp绿色荧光蛋白可以在荧光显微镜下直接观察,而常常作为一种标记基因;neo基因是一种抗生素抗性基因,细胞表达该基因可以灭活氨基糖甙类抗生素,破坏g418,而常常作为一种筛选标记基因用于转基因单克隆细胞的筛选。因而这两种基因无可避免的会出现在转基因动物中,而转基因动物成功获得后这些标记基因便不再需要了。
同时,外源标记基因的存在将会对转基因家畜的健康将产生不良的影响,且我国转基因安全评价政策明文规定将标记基因等同于目的基因,同样必须经过复杂的安全评价,这无疑直接影响了转基因家畜的推广和产业化进程。因此,在进行转基因的操作中,通常在标记基因的两侧各加上一个相同的loxp序列,以便在富集转基因成功的细胞后利用cre/loxp系统将转基因细胞内多余的标记基因删除。
猪圆环病毒,属于圆环病毒科,为单股环状dna病毒,分为1型和2型。其中猪圆环病毒1型(pcv1)不致病,猪圆环病毒2型(pcv2)具有致病性。在养猪业疾病防控中,疫苗是人们用来对付疾病的最常用手段。而由于圆环病毒灭活疫苗与亚单位疫苗成本高、注射次数多和容易造成机体应激等缺点,以及免疫效力有待提高,并不能大规模的免疫接种。另外,疫苗的使用在控制由pcv2感染而引起pmws中获得了一些成功,但是这种综合征的发病病源是多样的,且在其他综合征上并没有突破,所以通过转基因手段引入抗pcv2sirna基因序列,使用rna干扰技术对猪圆环病毒2型的防御显得更有实际价值。
在2013年12月25日公布的专利号为201310321355.6号,专利名称为“抗猪圆环病毒2型的表达载体和转基因猪及其构建方法”中公开了一种抗猪圆环病毒2型(pcv2)的表达载体和转基因猪及其构建方法,并通过该方法获得了f0代(原代)抗猪圆环病毒2型(pcv2)转基因猪,该转基因猪整合的外源基因包括目的基因抗pcv2sirna的基因序列及标记基因neo/egfp融合表达基因序列,而标记基因的存在,将对该转基因猪产生不良的影响,并影响该转基因猪的安全评价,不利于该转基因猪的推广及产业化进程。
技术实现要素:
本发明的目的是提供一种无外源标记基因的抗pcv2的转基因猪制备方法,以解决上述问题。
根据本发明的一个方面,提供了一种无外源标记基因的抗pcv2转基因猪制备方法,其是通过如下步骤来实现的:
(1)、抗pcv2表达载体的构建,该载体包含目的基因抗pcv2的sirna表达基因序列和标记基因序列;
(2)、筛选可稳定表达载体的单克隆转基因细胞系;
(3)、将获得的单克隆转基因细胞系作为核供体细胞,进行体细胞核移植获得f0代抗pcv2的转基因猪;
(4)、将获得的f0代抗pcv2的转基因猪与母猪杂交繁育获得能稳定遗传的抗pcv2的f1代转基因猪;
(5)、筛选f1代转基因猪个体并进行外源基因整合位点分析;
(6)、将筛选出的f1代转基因猪个体采取耳皮成纤维细胞,并进行耳皮成纤维细胞系的建立;
(7)、建立删除了外源标记基因的细胞单克隆,并进行体细胞克隆获得f2代无外源标记基因的抗pcv2的转基因猪。
由此,通过上述方法获得的抗pcv2的转基因猪,可以特异表达抗pcv2sirna,对猪圆环病毒疾病起到防疫作用;同时该转基因猪不含外源标记基因,不会对猪健康产生额外的负担及影响,同时也可以简化转基因猪安全评价程序,有利于该转基因猪的产业化推广及应用。
在一些实施方式中,上述外源标记基因是neo/egfp融合表达基因,neo基因是一种抗生素抗性基因,细胞表达该基因可以灭活氨基糖甙类抗生素,破坏g418,而常常作为一种筛选标记基因用于转基因单克隆细胞的筛选;而egfp为绿色荧光蛋白表达基因与目的基因共同转染构建转基因单克隆细胞系时,便于用显微镜直观的观察筛选结果,对转基因动物也可以通过观察绿色荧光蛋白的表达来初步判断转基因的结果,但egfp绿色荧光蛋白在细胞水平表达会对细胞产生一定的毒性,在转基因猪个体中也会对猪的健康产生一定的影响,因此删除了neo/egfp融合表达外源标记基因的转基因猪会减少对该转基因健康的影响,同时可以简化转基因猪安全评价程序,有利于该转基因猪的产业化推广及应用。
在一些实施方式中,f1代抗pcv2转基因猪是通过f0代抗pcv2转基因猪与同品种,且遗传背景一致的野生型母猪杂交繁育而来。
在一些实施方式中,f0代抗pcv2转基因猪与同品种,且遗传背景一致的野生型母猪杂交繁育而来的f1代抗pcv2转基因猪筛选的标准为高效特异表达抗pcv2sirna,且抗pcv2sirna基因序列的整合位点只有一个,由此筛选出来的f1代抗pcv2转基因猪既可以稳定遗传目的基因抗pcv2sirna,同时由于整合位点只有一个,便于定向删除与目的基因同时整合在转基因猪染色体上的外源标记基因。
在一些实施方式中,通过htncre重组酶来删除外源表达基因的步骤如下:
(1)、复苏f1代抗pcv2转基因猪耳皮成纤维细胞;
(2)、待细胞生长状态良好时,用htncre重组酶处理细胞系;
(3)、24h后,对处理后的细胞系进行传代培养,并在显微镜下挑选无绿色荧光表达的细胞单克隆团,并进行纯化培养,即获得无neo/egfp标记基因的细胞单克隆。
由此,通过htncre重组酶利用外源基因两侧的loxp位点,即可以删除外源标记基因,且删除标记基因后的细胞不表达绿色荧光,在细胞团中挑出不表达绿色荧光的细胞,进行纯化建系,即可以获得无neo/egfp标记基因的细胞单克隆。
在一些实施方式中,htncre重组酶处理细胞系删除外源标记基因时,具体要求如下:待f1代抗pcv2转基因猪耳皮成纤维细胞密度达80%左右时,用dpbs洗2次,加入opti-mem稀释的htncre重组酶,使其终浓度为200ng/μl,孵育3h后吸走htncre工作液,用dpbs洗2次,加入10%fbs的完全培养基继续培养细胞,由此可以获得最佳的删除效率,并获得能保证删除标记基因之后的细胞生长状态较好。
在一些实施方式中,挑选无绿色荧光表达的细胞单克隆团的具体步骤如下:当经htncre重组酶处理的细胞培养至10天左右,细胞克隆团细胞数达到200个左右时,在倒置显微镜的观察下于细胞培养皿底部圈出细胞克隆团所在位置,并在克隆团外围再圈出无杂细胞的安全区域,同时用荧光显微镜观察细胞克隆团有无荧光,做好标记,以确保挑取更纯的无荧光细胞单克隆。由此,可以精确的挑选出无绿色荧光蛋白表达的细胞团,建系培养后,保证细胞系中无未删除标记基因的杂细胞。
在一些实施方式中,获得无neo/egfp标记基因的细胞单克隆的具体步骤如下:标记好无荧光细胞单克隆后,吸走细胞完全培养基,用dpbs洗2次,用镊子将克隆环小心准确放在已做标记的细胞克隆上,每个克隆环内加入两滴0.25%胰酶,在显微镜下观察至细胞变圆时,加入50μl左右的fbs终止消化,用移液器小心吹起细胞并转移至24孔板中,按24孔-6孔-60mm的顺序进行传代培养,以获得无标记基因荧光表达的细胞单克隆。由此,可以保证细胞系无未删除标记基因的杂细胞,且保证筛选出的细胞系生长状态较好。
在一些实施方式中,删除外源标记基因时htncre重组酶的终浓度还可以为100ng/μl,150ng/μl或300ng/μl等浓度,只是优选的选用200ng/μl。
上述的无外源标记基因的抗pcv2转基因猪制备方法,具有如下有益效果:无外源标记基因的抗pcv2转基因猪是通过构建含有目的基因(抗pcv2sirna基因)及外源标记基因(neo/egfp融合表达基因)的共表达载体,进行转染制备转基因单克隆细胞系,然后以此单克隆细胞系作为体细胞克隆技术的供核体细胞,来获得含有目的基因及外源标记基因的f0代抗pcv2转基因猪,然后通过f0代与品种、遗传背景一致的野生型杜洛克母猪杂交繁育获得能稳定遗传的抗pcv2的f1代转基因猪,筛选出抗pcv2sirna表达量高,且只有一个整合位点的f1代转基因猪个体进行后续研究,采取该筛选出的f1代转基因猪个体的耳皮成纤维细胞进行培养建系,对获得的耳皮成纤维细胞系进行htncre重组酶处理,并筛选出删除了标记基因的细胞单克隆,以此细胞单克隆作为体细胞克隆技术的供核细胞,进行体细胞克隆获得f2代不含外源标记基因的抗pcv2转基因猪,并对该f2代转基因猪进行检测,证实标记基因已被删除,但抗pcv2sirna基因序列被保留。通过上述方法获得的无外源标记基因的抗pcv2转基因猪可以特异表达抗pcv2sirna,来抑制pcv2的表达及生物活性,从而起到防治猪感染圆环病毒疾病的作用。同时,该转基因猪删除了外源标记基因,减少了外源基因对转基因猪个体健康的影响,同时也可以简化转基因猪安全评价程序,有利于该转基因猪的产业化推广及应用。
附图说明
图1为实施例1中抗pcv2转基因猪f1代egfp外源标记基因绿色荧光表达情况;
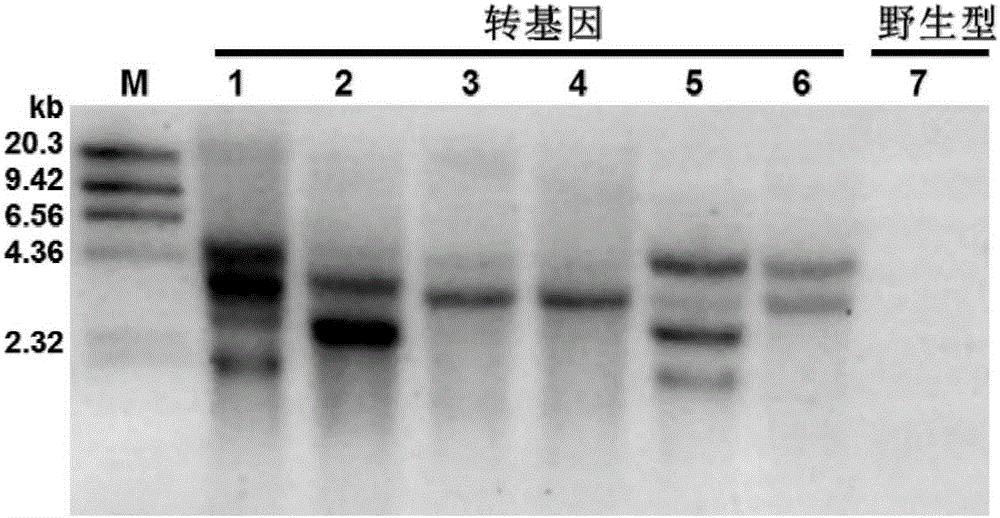
图2为实施例2中抗pcv2(转基因猪f1代的southernblot检测结果,其中mw为dna分子量标准,transgenic为f1代抗pcv2转基因猪样品,wild-type为野生型对照猪样品;
图3为实施例3中不同f1代抗pcv2转基因猪之间sirna相对表达情况,其中“3”标识tg3个体;
图4为实施例4中f1代抗pcv2转基因猪tg3个体目的基因整合位点测序对比图;
图5为实施例5中f1代抗pcv2转基因猪tg3耳皮成纤维细胞系的建立结果图;
图6为实施例6中htncre重组酶的表达纯化sds-page分析结果图,其中mw为蛋白分子量标准;1~3、5为诱导的溶菌产物粗分;4为未诱导的溶菌产物粗分;6为诱导的溶菌产物上清;7为诱导的溶菌产物沉淀;8为纯化后的产物;
图7为实施例7中htncre处理f1代抗pcv2转基因猪tg3细胞系前后流式细胞检测荧光结果图;
图8为实施例7中f1代抗pcv2转基因猪tg3细胞克隆删除标记前后荧光表达检测结果图;
图9为实施例8中删除标记的转基因猪tg3细胞单克隆的pcr检测结果图,其中mw为dnamarker;nagtive为阴性对照;w为水;wt为野生型猪;positive为阳性对照;p为pegfp-sh2质粒;-cre为未经htncre处理;mix为htncre处理后的混合细胞;c1~c7为7个细胞单克隆;
图10为实施例8中htncre重组酶介导的删除反应原理以及pcr产物(530bp)测序验证结果图;
图11为实施例10中无外源标记基因的抗pcv2转基因猪的egfp表达情况结果图;
图12为实施例11中无外源标记基因的抗pcv2转基因猪的pcr检测结果图。
具体实施方式
下面结合附图对发明作进一步详细的说明。
如无特殊说明,以下实施例中所使用的试剂均来源于市售,操作方法为现有常规操作方法。
本发明中所指的f1代抗pcv2转基因猪通过专利号为201310321355.6,专利名称为“抗猪圆环病毒2型的表达载体和转基因猪及其构建方法”所公布的方法获得的抗猪圆环病毒2型转基因克隆猪(f0代)与品种、遗传背景一致的野生型杜洛克母猪杂交繁育所得。且专利号为201310321355.6,专利名称为“抗猪圆环病毒2型的表达载体和转基因猪及其构建方法”所公布的抗猪圆环病毒2型转基因克隆猪(f0代)包含抗猪圆环病毒2型的sirna、筛选基因neo,绿色荧光蛋白标记基因egfp等外源基因,其中neo与egfp基因融合表达,该f0代抗猪圆环病毒2型转基因猪来源于本校。
实施例1:利用生物荧光灯观察f1代抗pcv2转基因猪的egfp表达。
在蓝色光激发条件下,肉眼透过相应的滤光片能观察到egfp蛋白所呈现的绿色荧光。基于此原理,利用生物荧光灯可以初步鉴定全身组织表达egfp蛋白的f1代抗pcv2转基因猪。具体操作如下:将转基因猪和野生型猪摆放在同一处,分别在相同的蓝色光背景和白光背景下观察并拍照。其中,蓝光背景下相机拍摄参数为:光圈优先,光圈f2.8,iso4000,手动对焦;白光背景下相机拍摄参数为:光圈优先,光圈f5.6,iso100,手动对焦。同理,对转基因猪和野生型猪解剖所取的部分器官组织进行观察并拍照。
检测结果如附图1所示,在f1代抗pcv2转基因猪的口鼻部位以及器官组织肾、肺、心均能观察到明显的绿色荧光表达,而阴性对照在同样条件下未能观测到绿色荧光。这表明,f1代抗pcv2转基因猪能稳定遗传并高效表达整合到f0代转基因猪基因组的外源基因。
实施例2:利用southernblot检测外源基因在f1代抗pcv2转基因猪中的整合。
通过southernblot可以鉴定外源基因在转基因猪基因组中整合的拷贝数,其原理大致为:先对pegfp-sh2载体序列进行酶切位点分析,选择一个比较合适的单酶切位点(hindiii),然后用pegfp-sh2质粒设计1拷贝数的对照,用f0代抗pcv2转基因猪基因组dna作为阳性对照,并以哺乳动物基因组dna大小约为3×109个碱基对(bp)作为依据,按下述公式计算拷贝对照所需要的质粒量:
质粒量(μg)=插入质粒片段大小(bp)/3*109(bp)×基因组dna量(μg)
从而根据结果估计f1代抗pcv2转基因猪插入外源基因的拷贝数。具体操作步骤如下:
(1)pcr法制备southernblot所需探针
southernblot所需探针采用pcr标记法制备,其原理为:在表达目的sirna的质粒片段两端分别设计引物,采用pcrdigprobesynthesiskit来标记,扩增得到的650bppcr产物即为标记好的探针。引物序列及扩增产物长度见表如下:
反应体系的配制如下:
具体操作步骤为:
1)将试剂各组分于冰上解冻,短离心,混匀;
2)按上表中各组分的量由大体积到小体积依次加入反应管中,所有操作均在冰上进行,混匀,短离心;
3)将反应管置于pcr仪中进行扩增反应,程序如下:95℃-2min;95℃-30s,60℃-30s,72℃-40s;30cycles;72℃-7min;
4)检测探针标记效率:吸取等体积的pcr产物上样于2%的琼脂糖凝胶进行电泳,如果可见目的条带大小正确,并且dig标记探针比未标记对照探针迁移速度稍慢,则表明所标记探针制备成功,-20℃保存备用
(2)southernblot杂交流程
southernblot所用试剂盒为dighighprimednalabelinganddetectionstarterkitii,具体操作步骤根据试剂盒说明书进行。
检测结果如附图2所示,表明检测的6头f1代抗pcv2转基因猪存在1-4个整合位点,其中tg3和tg4均只有一个整合位点,而野生型对照猪则没有获得southernblot结果,表明本实验所制备的探针是特异的,f1代抗pcv2转基因猪特异地整合了目的基因及外源标记基因。
实施例3:利用real-timeqpcr检测sirna在f1代抗pcv2转基因猪中的表达。
(1)总rna提取
rna组织样浸泡于rnalater中并保存于-80℃。应用mirvanamirnaisolationkit提取总rna,具体操作步骤根据说明书进行。
(2)特定sirna的反转录
根据目的基因sirna和内参基因mir16序列设计引物,用同一rna模板分别于pcr管中进行反转录反应,经引物扩增后得到cdna。引物序列如下:
反转录(7.5μl反应体系)mastermix体系的配制如下:
反转录mastermix体系的配制过程如下:
i.轻轻混匀,切勿涡旋,离心至管底,然后放置于冰上;
ii.对于每一个7.5μl的反应体系,加入1μl5ng~10ng的总rna;
iii.轻轻混匀,切勿涡旋,短离心至管底,冰上操作;
iv.加入3μl50nm的引物,引物使用前混匀并短离心;
v.混匀,切勿涡旋,短离心至管底,冰上孵育5min;
vi.反转录程序为:16℃,30min;42℃,30min;85℃,5min;4℃保存。
(3)特定sirna的real-timeqpcr检测
i.目的基因sirna及内参基因mir16的qpcr检测引物及探针如下:
ii.sirnaqpcr20×taqmansirnaassay100μl体系的配制如下:
iii.sirnaqpcr反应体系的配制如下:
上述试剂均在冰上溶解,混匀,短离心至管底。qrcr反应程序为:95℃,10min;95℃,15s,60℃,1min;40cycles。
检测结果如附图3所示,结果表明不同f1代转基因猪之间sirna的表达存在差异,其表达量与southernblot结果中得知的外源目的基因拷贝数多少并不严格对应,推测这可能与外源目的基因在基因组上的整合位置有关;而外源基因均为单拷贝的个体tg3的表达量是tg4的4倍左右。
根据实施例2和实施例3的结果表明,tg3转基因猪个体在外源基因只有一个整合位点,且具有较高的抗pcv2sirna表达量,因而选择筛选出该tg3个体进行后续研究。
实施例4:利用ipcr(反向pcr)确定外源基因在tg3抗pcv2转基因猪基因组中的整合位点。
经过southernblot初步判断外源基因在tg3个体基因组中整合情况后,进一步采用ipcr的方法寻找外源基因的整合位点,一方面可以验证southernblot的结果,另一方面可以分析外源基因在染色体上的整合位置对转基因猪的影响,另一方面还可以为删除标记基因做准备。通过分析southernblot的结果,可以判断hindiii限制性内切酶同样适用于ipcr。ipcr具体操作步骤如下:
(1)基因组dna的提取
参照实施例2中的步骤。
(2)基因组dna的酶切及纯化
参照实施例2中的步骤,dna用量为2μg。
(3)酶切产物的连接及纯化
取上述酶切回收的dna样品0.2μg~2μg置于0.5ml离心管中,再依次加入连接反应试剂,混匀,短离心,将反应管置于16℃恒温金属浴中反应12h-16h,最后纯化回收,具体纯化步骤如上。
(4)ipcr检测
ipcr以纯化回收的连接产物为模板进行反应,所用引物序列如下:
以上4条引物序列中,其中pb3’treprimary和cmvprimary为一对,用于扩增转座子3’端和整合位点附近的基因组序列;pb5’treprimary和neoprimary为一对,用于扩增转座子5’端和整合位点附近的基因组序列。
(5)ipcr产物切胶回收
将ipcr产物全部进行琼脂糖凝胶电泳,使各条带尽可能地分开以便切胶回收。切胶回收采用e.z.n.a.gelextractionkit,具体操作步骤根据说明进行。
(6)t-a克隆
经切胶回收的ipcr产物3’末端带有a碱基,能直接与instaclonepcrcloningkit中的t-vector进行t-a克隆反应,反应体系如下:
将上述混合液混匀,短离心,至于22℃反应1h,连接后的产物用来转化dh5α感受态细胞。
(7)转化
i.将保存于-80℃的感受态细胞取出置于冰上解冻;
ii.将50μl感受态细胞移至灭菌处理的离心管内,加入上述连接产物,轻轻混匀,冰浴30min;
iii.42℃热击45s~60s,然后迅速转移至冰中放置2min,此过程不要摇动离心管;
iv.加入500μl37℃预热的lb培养基,置于恒温空气摇床,37℃,220rpm,复壮1h;
v.取适量复壮后的菌液涂布于经iptg和x-gal处理的la固体培养基,并将其置于37℃至液体被吸收,倒置,37℃培养12h~16h。
(8)蓝白斑筛选及菌液pcr鉴定
在上述la培养板上挑取白色单克隆菌落,置于1mlla液体培养基中,37℃,220rpm,培养4h;利用ipcr检测引物进行菌液pcr鉴定。(9)菌液测序及比对分析
将上述pcr鉴定为阳性的单克隆菌液送往华大基因进行测序,并将成功测序的序列于ncbi上与pegfp-sh2载体序列及猪全基因组序列进行比对,分析寻找整合位点。
检测结果如附图4所示,测序及比对结果表明在tg3个体中,外源基因整合在第10号染色体上,位于第10号染色体两个功能基因cugbpelav-likefamilymember与trans-actingt-cell-specifictranscriptionfactorgata-3之间,不会影响该tg3转基因猪正常基因的表达,同时外源基因整合在该位点具有较高的表达效果。
实施例5:tg3抗pcv2转基因猪的耳皮成纤维细胞系的建立。
tg3抗pcv2转基因猪出生后,经过pcr和southernblot鉴定,确定为后续需要深入研究的个体,采其耳皮组织,在实验室进行耳皮成纤维细胞建系,方法如下:
(1)用碘酊对要取样的部位以及耳缺钳、镊子、等进行消毒,尽可能地除去耳皮表面的污垢,剪取一块小指指甲般大小的组织块,置于碘酊中泡1min~2min,然后转移至75%乙醇消毒液进行脱碘并消毒约30s,再用镊子夹住组织于含2%双抗的dpbs中涮洗3次,最后快速放入装有含2%双抗的无血清dmem的50ml离心管中盖紧,置于冰盒中尽快带回实验室进行下一步的处理,以保证耳皮细胞的活性;
(2)将组织从管中取出置于细胞培养皿中,用含2%双抗的dpbs洗涤3~5次,剪除毛茬,用手术刀片轻轻刮去表皮细胞,保留真皮层,再将组织泡于碘酊30s,75%乙醇消毒液脱碘30s,含2%双抗的dpbs涮3~5次,用眼科剪将耳皮组织剪碎成小块;
(3)用完全培养基清洗组织碎末2次后用1ml的血清重悬,再转移到新的培养皿中并使其均匀分布,放入37℃、5%co2及饱和湿度的细胞培养箱中,待培养5h~7h至组织小块贴附于培养皿底后添加完全培养基;
(4)每隔2天换液一次并观察组织块周围细胞爬出情况,待细胞长至80%~90%汇合度时进行传代培养和冻存。
检测结果见附图5,结果表明tg3耳皮成纤维细胞系成功建系的细胞贴壁正常,生长速度良好,呈长梭形或不规则形,富有立体感,有较强的绿色荧光表达。
实施例6:htncre重组酶的表达纯化。
(1)将htncre重组酶原核表达质粒转化于bl21(de3)菌株,37℃培养过夜。在平板上挑单克隆接入3ml的la液体培养基中(含50μg/mlamp),37℃,250rmp振荡培养至od600=0.5~0.6后,(a)加入0.5mmiptg37℃诱导3h;(b)加入0.1mmiptg16℃诱导过夜;sds-page检测表达。
(2)重组质粒转化的bl21(de3)表达菌株接种于100mlla液体培养基中,37℃培养过夜,第二天转接到1lla液体培养基中,37℃,250rmp振荡培养至od600=1.0后,加入0.1mmiptg16℃诱导过夜,收菌。
(3)sp-ffcolumn用20mmpb,200mmnacl,1mmedta,ph8.0缓冲液平衡,以20mmpb,1mnacl,1mmedta,ph8.0缓冲液梯度洗脱,收集洗脱组分。
(4)superdex200column以pbs,10%glycerol,ph7.4缓冲液平衡,sp-ffcolumn洗脱收集组分上样后,以pbs,10%glycerol,ph7.4缓冲液分步洗脱,收集洗脱组分,浓缩得到蛋白产品,检测蛋白浓度。
检测结果如附图6所示,从附图6中的sds-page凝胶电泳检测可见,诱导菌株bl21(de3)过量表达产物在分子量大小约为40kda处出现表达量相对较高的明显条带,与理论计算的大小42.76kda相符,初步判断该条带为目的重组蛋白条带。本实验获得可溶性表达,但表达不明显,经过大量收集溶菌产物上清,最后从上清中浓缩纯化得可溶性htncre重组酶1.2mg,浓度为0.6mg/ml,储存于pbs、10%glycerol、ph7.4缓冲液中,小管分装后于-80℃长期保存备用,尽量避免反复冻融。
实施例7:利用htncre删除tg3个体耳皮成纤维细胞系neo/egfp外源标记基因。
(1)复苏tg3个体耳皮成纤维细胞于60mm细胞培养皿,长满后传代至6孔板,培养条件为37℃,5%co2。
(2)待细胞密度达80%左右时,用dpbs洗2次,加入opti-mem稀释的htncre,使其终浓度为200ng/μl,孵育3h后吸走htncre工作液,用dpbs洗2次,加入10%fbs的完全培养基(含1%双抗)继续培养。
(3)24h后,用0.25%的胰酶消化细胞,用细胞计数仪对细胞进行计数,按200个细胞/皿传至100mm细胞培养皿中,每隔3天换一次液并观察细胞单克隆的形成情况。
(4)当细胞培养至10天左右,细胞克隆团细胞数达到200个左右时,用marker笔在倒置显微镜的观察下于细胞培养皿底部圈出细胞克隆团所在位置,并在克隆团外围再圈出无杂细胞的安全区域,同时用荧光显微镜观察细胞克隆团有无荧光,做好标记,以确保挑取更纯的无荧光细胞单克隆。
(5)吸走完全培养基,用dpbs洗2次,用镊子将克隆环小心准确放在已做标记的细胞克隆上,每个克隆环内加入两滴0.25%胰酶,在显微镜下观察至细胞变圆时,加入50μl左右的fbs终止消化,用移液器小心吹起细胞并转移至24孔板中,按24孔-6孔-60mm的顺序进行传代培养,可在传代过程中一分为二,一份用于pcr鉴定,一份冻存备用。每传一代都需进行荧光观察,进一步确保筛选到的细胞单克隆中无未删除荧光标记的杂细胞。
检测结果用实施例6中表达纯化的htncre重组酶处理f1代抗pcv2转基因猪tg3耳皮成纤维细胞系,由于绿色荧光蛋白在htncre重组酶处理前就已经存在,在htncre删除标记之后其代谢需要一定的时间,所以细胞系标记被删除的效果在24h内并不能明显观察到,细胞始终有可见的绿色荧光。但随着细胞的继续分裂生长,绿色荧光越来越弱。如附图7所示的是,tg3细胞系在htncre处理里后第7天进行流式细胞分析,发现tg3表达绿色荧光的细胞由标记未删除前的99.8%变为删除标记后的4.8%,标记删除效率高达95%。
检测结果如附图8所示:htncre处理f1代抗pcv2转基因猪tg3耳皮成纤维细胞系后,稀释培养并用克隆环进行细胞单克隆筛选,发现未经过htncre删除标记的细胞单克隆表达较强的绿色荧光(40×),而经过htncre删除标记的则未能观察到有绿色荧光表达。
同时,由于标记基因neo与绿色荧光蛋白基因egfp为融合表达,且位于两个loxp之间,因而egfp被删除的同时,neo基因也会被同时删除。
在其他实施例中,htncre重组酶浓度还可以为100ng/μl,150ng/μl或300ng/μl等浓度,均可以具有较高效率的删除外源标记基因的效果,只是优选的采用200ng/μl时,删除效果最好。
实施例8:利用pcr检测删除标记基因的细胞单克隆。
(1)细胞dna提取参照实施例2中的步骤;
(2)利用pcr检测neo/egfp融合表达标记基因和两个loxp之间的序列,所用引物如下:
用pcr对筛选到的f1代抗pcv2转基因猪tg3细胞单克隆的neo/egfp标记以及两个loxp之间的标记序列进行检测,如附图9所示,筛选到的c1~c7无绿色荧光细胞单克隆均未检测到neo/egfp标记以及loxp之间的标记序列,而阳性对照中的质粒、未处理以及处理后的混合细胞中则均能检测到,说明筛选到的细胞单克隆中的标记已被htncre重组酶介导的删除反应删除掉,其删除反应原理以及删除标记后的pcr产物测序验证见附图10,即标记删除前能扩增到980bp的neo/egfp标记以及3211bp的两个loxp之间的序列,而标记删除后扩增不到980bp的neo/egfp标记以及只能扩增到530bp两个loxp之间序列被删除后剩下的片段。
实施例9:利用scnt制备无neo/egfp标记基因的抗pcv2转基因克隆猪。
将实施例7中获得的无neo/egfp标记基因的tg3个体耳皮成纤维细胞系作为核供体细胞,注射到卵母细胞中,构建重构胚胎进行体细胞克隆(scnt),并采用常规育种技术进行培育,即可获得抗pcv2的转基因猪。实施例10:利用生物荧光灯检测无neo/egfp标记基因抗pcv2转基因克隆猪的egfp表达。
检测方法同实施例1。
用生物荧光灯对获得的无neo/egfp标记基因抗pcv2转基因克隆猪进行egfp表达检测,选择大小相似的表达egfp的猪作为对照,对照猪可见较明显的egfp表达,尤其是在口蹄部,而无neo/egfp标记抗pcv2转基因克隆猪则观察不到egfp的表达,如附图11,判断通过实施例9获得的无neo/egfp标记抗pcv2转基因克隆猪其egfp标记被删除,由于neo基因与egfp基因融合表达,egfp基因不表达时,neo基因也不会表达。
实施例11:利用pcr检测无neo/egfp标记基因的抗pcv2转基因克隆猪。
检测方法同实施例8。经pcr进一步检测,检测结果如附图12,结果表明,实施例9中获得的抗pcv2转基因小猪均成功删除neo/egfp外源标记基因,而目的基因抗pcv2sirna基因序列予以保留,也进一步证明筛选用于制备无标记抗pcv2转基因克隆猪的细胞单克隆较纯且成功删除了标记基因,与绿色荧光检测结果相符合。表明通过上述方法获得了无外源neo/egfp标记基因的抗pcv2转基因克隆猪。
以上所述的仅是本发明的一些实施方式。对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 化药/基因共转运功能化碳纳米管的制备方法及应用与流程
- 抗体基因的表达‑分泌系统的制作方法与工艺
- 抗体捕获蛋白及报告基因融合重组蛋白检测抗体的方法与流程
- 治疗性重组抗体、其编码基因及应用的制作方法与工艺
- 一种制备高效合成泛酸基因工程菌的方法及其应用与流程
- MACF1基因及其表达产物在制备诊断和治疗骨质疏松症药物中的应用的制作方法与工艺
- 野生型IDH2基因作为靶标在制备非小细胞肺癌治疗药物中的应用的制作方法与工艺
- FBN1基因在制备检测先天性单纯性晶状体异位制品中的应用的制作方法与工艺
- TMEM104基因在制备治疗椎间盘退行性疾病药物中的应用的制作方法与工艺
- PSMG4基因在制备男性骨质疏松症诊断产品中的用途的制造方法与工艺