替吉奥杂质BCB的制备方法与流程
本发明属于有机化学合成领域,具体涉到一种替吉奥杂质bcb的制备方法,所述杂质bcb化学名称为3,3'-(4-羟基丁烷基-1,1-二基)双(5-氯吡啶-2,4-二醇)。
背景技术:
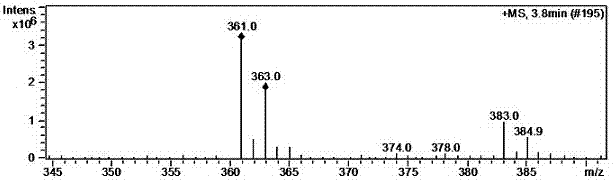
:替吉奥胶囊是一种氟尿嘧啶衍生物口服抗癌剂,它包括替加氟(ft)和以下两类调节剂:吉美嘧啶(cdhp)及奥替拉西(oxo)。替吉奥往往含有杂质3,3'-(4-羟基丁烷基-1,1-二基)双(5-氯吡啶-2,4-二醇)(bcb),该杂质是替吉奥制剂不可避免的杂质,也是替吉奥制剂中必须标定的杂质项目之一。在替吉奥胶囊制剂的进口质量标准中,明确提到bcb作为特定杂质来控制。为了标定替吉奥制剂中杂质bcb,需首先获得纯的bcb作为对照品,但是在替吉奥胶囊中各组分的合成及相关药理毒理文献中,对bcb的报导非常少,只有只言片语提到bcb,而关于bcb的合成,没有任何文献专利论文报导其合成。技术实现要素:替吉奥胶囊是一种氟尿嘧啶衍生物口服抗癌剂,它包括替加氟(ft)和以下两类调节剂:吉美嘧啶(cdhp)及奥替拉西(oxo)。替吉奥往往含有杂质3,3'-(4-羟基丁烷基-1,1-二基)双(5-氯吡啶-2,4-二醇)(bcb),该杂质是替吉奥制剂不可避免的杂质,也是替吉奥制剂中必须标定的杂质项目之一。在替吉奥胶囊制剂的进口质量标准中,明确提到bcb作为特定杂质来控制。为了标定替吉奥制剂中杂质bcb,需首先获得纯的bcb作为对照品,但是在替吉奥胶囊中各组分的合成及相关药理毒理文献中,对bcb的报导非常少,只有只言片语提到bcb,而关于bcb的合成,没有任何文献专利论文报导其合成。鉴于bcb在替吉奥质量控制中的关键地位,本发明在详细研究了原料药中各组分以及制剂处方工艺基础上,提出了一种高效制备高纯度bcb的方法。为替吉奥胶囊的质量控制提供杂质对照品。本发明是通过如下技术方案实现的:一种替吉奥杂质bcb的制备方法,所述吉奥胶囊杂质bcb为3,3'-(4-羟基丁烷基-1,1-二基)双(5-氯吡啶-2,4-二醇)(bcb),其结构式为,反应路线如下所示:。其中,式ii为吉美嘧啶,式iii为替加氟,式ⅰ为bcb,式ⅳ为5-氟尿嘧啶。上述的替吉奥杂质bcb的制备方法,包括如下步骤:(1)吉美嘧啶与替加氟在催化剂催化下升温至40~75℃,搅拌条件下保温反应60~90h,反应结束后,将反应体系中的反应溶剂浓缩除掉,得到固体,然后进行过滤,并用水洗涤滤饼,将滤饼转移至反应瓶中,加入二氯甲烷回流0.5小时,在进行过滤,收集滤饼,得到含有bcb和吉美嘧啶的混合物;(2)将步骤1得到的混合物进行柱层析,选用200-300目的硅胶,硅胶的用量为样品重量的50倍,以醋酸和乙酸乙酯混合溶剂作为洗脱剂,柱层析即得产物bcb。所述步骤(1)中吉美嘧啶与替加氟的投料摩尔比为1:2.1~2.5。优选为1:2.2。所述步骤(1)中的反应溶剂选自甲醇、乙醇、水、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺或二甲基亚砜中的两种或多种。所述步骤(1)中吉美嘧啶与替加氟的反应时间为72~90h。所述步骤(1)中吉美嘧啶与替加氟的反应温度为50~60℃。所述步骤(2)中洗脱剂为乙酸乙酯和醋酸混合溶剂,乙酸乙酯与醋酸体积比为30~60:1。优选为为50:1。由bcb的结构分析可知,分子结构对称,有两个吉美嘧啶结构,连在吉美嘧啶结构中间的是正丁醇的结构。替加氟结构中,与氮相连的是呋喃环结构,如果替加氟的碳氮键断裂的话,断裂的呋喃环与吉美嘧啶反应,有可能生成正丁醇结构。因此,发明人推测此反应的反应机理如下:反应的收率并不是制备杂质对照品的重点,此反应也不是一个完全转化的反应,利用hplc检测反应液,面积归一化法大约20%左右。如何纯化得到高纯度的bcb更为关键。诚然,利用制备液相分离固然可以提纯,那样会浪费大量的流动相,费时费力,且成本非常高。发明人仔细分析了反应中各物质的性质及转化的副产物后,通过简单的后处理,摸索出了简单高效的提纯方法。反应体系中主要含有替加氟、吉美嘧啶、bcb以及替加氟的分解产物5-氟尿嘧啶(iv)。上述物质中,5-氟尿嘧啶水溶性较好,bcb和替加氟在水中几乎没有溶解度。尽可能让替加氟分解成5-氟尿嘧啶,让其在水中溶解,可以过滤后进入水相除去。将得到的滤饼主要含有bcb和吉美嘧啶的混合物干燥后,再用二氯甲烷加热回流,bcb和吉美嘧啶在二氯甲烷中不溶,将其它溶于二氯甲烷的其它杂质除去。此时得到的混合物,经hplc分析只含有bcb和吉美嘧啶,然后经过一次柱层析分离即可得到高纯的bcb杂质。本发明的制备方法,工艺简单,反应条件温和,可以得到高纯度的产品bcb。附图说明图1为实施例1制备的bcb的h-nmr图谱。图2为实施例1制备的bcb的质谱图谱。图3为实施例1制备的bcb的高效液相色谱。具体实施方式:下面结合具体实施例对本发明作更进一步的说明,以便本领域的技术人员更了解本发明,但并不因此限制本发明。实施例1起始原料分子量投料量(g)替加氟200.1720吉美嘧啶14529氢氧化钠401乙醇46180水18120500ml的三口瓶中,依次加入20g替加氟、29g吉美嘧啶以及1g的氢氧化钠,再加入180g乙醇和120g水,开启搅拌,升温至55℃,溶液为澄清透明体系,保温反应75h。反应结束后,40℃下,减压浓缩,蒸除乙醇,将200g水加入到剩余物中,然后升温至60℃继续反应4h。反应结束后,继续降温至室温,过滤,收集滤饼。50℃鼓风烘箱干燥8h,得35g粉末状固体。将上述35g固体加入到300ml二氯甲烷中,加热回流2h,降至室温后,过滤,收集滤饼,50℃鼓风干燥2h,得固体约34g。取少量固体,甲醇溶解,tlc检测,(展开剂为乙酸乙酯:醋酸=50:1),碘缸显色,固体只含有bcb和吉美嘧啶。将上述固体硅胶柱层析分离,选用200~300目的硅胶,硅胶的用量为样品重量的50倍,洗脱剂为乙酸乙酯和醋酸,其中乙酸乙酯:醋酸=50:1(体积比)。柱层析分离得到bcb烘干后称重约3.8g,收率10.5%,hplc检测纯度99.75%。下表为制备的bcb的液相hplc数据,其中分析结果如下:图1所示为制备的bcb的氢谱谱图,1h-nmr数据如下:化学位移δ质子数1.28522.25823.34624.39617.706212.315214.3061图2所示为制备的bcb的质谱图谱,其结果为:m/z=361。图3所示为制备的bcb的高效液相色谱,图谱中可见按照面积归一化法计算,本发明实施例1制备的bcb杂质纯度为99.75%。实施例2起始原料分子量投料量(g)替加氟200.1710吉美嘧啶14514.5氢氧化钠400.5乙醇4690水1860250ml的三口瓶中,依次加入10g替加氟、14.5g吉美嘧啶以及0.5g的氢氧化钠,再加入90g乙醇和60g水,开启搅拌,升温至75℃,溶液为澄清透明体系,保温反应90h。反应结束后,40℃下,减压浓缩,蒸除乙醇,将100g水加入到剩余物中,然后升温至60℃继续反应4h。反应结束后,继续降温至室温,过滤,收集滤饼。50℃鼓风烘箱干燥8h,得16.5g粉末状固体。将上述16.5g固体加入到150ml二氯甲烷中,加热回流2h,降至室温后,过滤,收集滤饼,50℃鼓风干燥2h,得固体约15.8g。取少量固体,甲醇溶解,tlc检测,(展开剂为乙酸乙酯:醋酸=50:1),碘缸显色,固体只含有bcb和吉美嘧啶。将上述固体硅胶柱层析分离,选用200~300目的硅胶,硅胶的用量为样品重量的50倍,洗脱剂为乙酸乙酯和醋酸,其中乙酸乙酯:醋酸=50:1(体积比)。柱层析分离得到bcb烘干后称重约1.25g,收率6.9%,hplc检测纯度99.45%。实施例3起始原料分子量投料量(g)替加氟200.1710吉美嘧啶14514.5氢氧化钠400.5乙醇4690水1860250ml的三口瓶中,依次加入10g替加氟、14.5g吉美嘧啶以及0.5g的氢氧化钠,再加入90g乙醇和60g水,开启搅拌,升温至40℃,溶液为澄清透明体系,保温反应80h。反应结束后,40℃下,减压浓缩,蒸除乙醇,将100g水加入到剩余物中,然后升温至60℃继续反应4h。反应结束后,继续降温至室温,过滤,收集滤饼。50℃鼓风烘箱干燥8h,得18.5g粉末状固体。将上述18.5g固体加入到150ml二氯甲烷中,加热回流2h,降至室温后,过滤,收集滤饼,50℃鼓风干燥2h,得固体约17.8g。取少量固体,甲醇溶解,tlc检测,(展开剂为乙酸乙酯:醋酸=50:1),碘缸显色,固体只含有bcb和吉美嘧啶。将上述固体硅胶柱层析分离,选用200~300目的硅胶,硅胶的用量为样品重量的50倍,洗脱剂为乙酸乙酯和醋酸,其中乙酸乙酯:醋酸=50:1(体积比)。柱层析分离得到bcb烘干后称重约0.6g,收率3.3%,hplc检测纯度98.75%。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1