一种苯硼酸功能化两性离子嵌段共聚物和葡萄糖敏感仿生纳米载体的制作方法
本发明涉及高分子材料技术领域,尤其涉及一种苯硼酸功能化两性离子嵌段共聚物和葡萄糖敏感仿生纳米载体。
背景技术:
糖尿病患者日益年轻化且患者人数急剧增加严重危害人类健康,对于糖尿病的治疗也已迫在眉睫。糖尿病是一种内源性疾病,是由于体内胰岛素分泌不足而造成的血糖升高尿中含糖的一种病症。常规注射胰岛素虽能快速降低血糖,但频繁注射带给病人带来极大的痛苦,病人的耐受性较差。
环境敏感型材料是能够响应外界环境刺激,如温度、离子浓度、ph值、葡萄糖等,并发生相应变化的一类高分子材料。其中,葡萄糖敏感药物载体由于能够响应葡萄糖浓度的变化而发生相应性能的改变,从而将所担载的糖尿病治疗药物释放出来而得到广泛的研究。
基于苯硼酸(pba)的葡萄糖敏感胰岛素自调式给药材料受到广泛关注。苯硼酸及其衍生物能够与多醇基化合物发生成酯反应生成苯硼酸酯。苯硼酸功能化纳米药物载体能够与小分子葡萄糖形成苯硼酸酯,一方面苯硼酸酯更加亲水,增加了纳米药物载体的亲水性,另一方面平面三角形的苯硼酸转化为正四面体的苯硼酸酯增大了纳米药物载体的空间体积。两方面的作用均使得纳米药物载体响应葡萄糖的刺激而释放出药物。基于苯硼酸pba的葡萄糖敏感材料在治疗糖尿病的研究中具有更广阔的前景。
现有技术公开了多种含有苯硼酸的葡萄糖敏感纳米载体材料,如南开大学的李朝兴教授课题组制备的糖基化聚合物聚(甲基丙烯酸2-(n-葡萄糖酰胺)乙酯-r-丙烯酰胺基苯硼酸)(p(gama-r-aapba))能够自组装成纳米粒子。该纳米粒子具有在糖尿病患者血糖水平快速释放胰岛素而在正常血糖水平缓慢释放胰岛素的特性,并具有良好的降血糖效果(polymerchemistry.,2016,7(18):3189-3199);现有技术还公开了一种采用海藻酸钠接枝聚(l-谷氨酸-co-n-3-l-谷氨酰胺基苯硼酸)(sa-pgga)和改性壳聚糖(gc)设计的新型聚氨基酸双层纳米凝胶(rscadvance,2015,5(19):14482-14491)。上述葡萄糖敏感纳米载体对葡萄糖浓度均较为敏感并采用了可生物降解类材料具有较好的生物相容性。但现有技术制备的纳米药物载体并未采用两性离子聚合物载体,使其应用领域受到限制。
技术实现要素:
有鉴于此,本发明的目的在于提供一种苯硼酸功能化两性离子嵌段共聚物和葡萄糖敏感仿生纳米载体,本发明提供的葡萄糖敏感仿生纳米载体具有良好的生物降解性、生物相容性和葡萄糖敏感性。
本发明提供了一种苯硼酸功能化两性离子嵌段共聚物,具有式i结构:
式i中,r1为h或ch3;
r2为磷酰胆碱基团、羧酸甜菜碱基团或磺酸甜菜碱基团;
k为聚合度,20≤k≤500;
n为聚合度,20≤n≤500;
0.1≤j/n≤1;
0≤i/n≤0.9。
在本发明中,r2优选为下式结构:
在本发明中,式a中与o连接的断键在r2的位置处与c键合;与n连接的未给出的端基均为甲基。
在本发明中,式b中与o连接的断键在r2的位置处与c键合;与n连接的未给出的端基均为甲基。
在本发明中,式c中与o连接的断键在r2的位置处与c键合;与n连接的未给出的端基均为甲基。
在本发明中,式d中与nh连接的断键在r2的位置处与c键合;与n连接的未给出的端基均为甲基。
在本发明中,k优选为100~200,更优选为110~150;100≤k≤200,更优选为110≤k≤150。在本发明中,n优选为30~300,更优选为40~100。在本发明中,j/n优选为0.1~0.7,更优选为0.2~0.7。在本发明中,i/n优选为0.1~0.7,更优选为0.3~0.6,最优选为0.3~0.5。
在本发明中,式i中聚合度j的聚合单元与nh连接的未给出的端基可以为h。在本发明中,式i中聚合度i和聚合度j聚合单元之间的/表示两聚合单元的聚合方式为无规共聚。
在本发明中,所述式i结构的苯硼酸功能化两性离子嵌段共聚物的数均分子量优选为9500~276000,更优选为24700~130100,最优选为30600~81000。
本发明提供了一种上述技术方案所述的苯硼酸功能化两性离子嵌段共聚物,包括以下步骤:
将具有式ii结构的聚合物和氨基苯硼酸在缩合剂的作用下反应,得到苯硼酸功能化两性离子嵌段共聚物:
式ii中,r1为h或ch3,
r2为磷酰胆碱基团、羧酸甜菜碱基团或磺酸甜菜碱基团;
k为聚合度,20≤k≤500;
n为聚合度,20≤n≤500。
在本发明中,聚合度n的聚合单元与nh连接的未给出的端基可以为h。
在本发明中,所述缩合剂优选包括1-(3-二甲基氨基丙基)-3-乙基碳化二亚胺盐酸盐和n-羟基琥珀酰亚胺;n-羟基琥珀酰亚胺先对羧基活化,然后1-(3-二甲基氨基丙基)-3-乙基碳化二亚胺盐酸盐再将活化的羧基和氨基反应脱去一份子水生成酰胺键。
在本发明中,所述的苯硼酸功能化两性离子嵌段共聚物的制备方法优选为:
将式ii结构的聚合物、1-(3-二甲基氨基丙基)-3-乙基碳化二亚胺盐酸盐和n-羟基琥珀酰亚胺置于小安瓶中,加入溶剂溶解上述混合物;进行活化反应,然后加入氨基苯硼酸进行缩合反应,制备得到苯硼酸功能化两性离子嵌段共聚物。
在本发明中,所述溶剂优选为n,n-二甲基甲酰胺。本发明对所述溶解的方法并无限制,采用本领域技术人员熟知的溶解方法即可。
在本发明中,所述活化反应温度优选为室温,如20~30℃,更优选为22~28℃,最优选为24~26℃;所述活化反应时间优选为45~55h,更优选为48h。在本发明中,所述缩合反应的时间优选为65~75h,更优选为72h。
本发明优选将具有式ii结构的聚合物用溶剂溶解,加入1-(3-二甲基氨基丙基)-3-乙基碳化二亚胺盐酸盐和n-羟基琥珀酰亚胺进行活化反应。
在本发明中,所述1-(3-二甲基氨基丙基)-3-乙基碳化二亚胺盐酸盐与式ii结构的聚合物中羧基的摩尔比优选为(1~5):1,更优选为(1~3):1。
在本发明中,所述活化反应温度优选为室温,所述活化反应的时间优选为1~2天。
在本发明中,所述活化反应完毕后,向得到的反应溶液中加入氨基苯硼酸进行缩合反应,得到式i结构的苯硼酸功能化两性离子嵌段共聚物。在本发明中,所述缩合反应的温度优选为0℃~20℃,更优选为5~15℃;所述缩合反应的时间优选为20h~60h,更优选为30~50h,最优选为35~45h。
在本发明中,所述式ii结构的聚合物的数均分子量优选为7000~222000,更优选为25000~102000,最优选为29000~66800。
在本发明中,所述氨基苯硼酸可以为邻位氨基苯硼酸、间位氨基苯硼酸或对位氨基苯硼酸,如2-氨基苯硼酸、3-氨基苯硼酸或4-氨基苯硼酸,优选3-氨基苯硼酸。
在本发明中,所述式ii结构的聚合物中羧基基团和氨基苯硼酸中氨基基团的摩尔比优选为1:(1~30),更优选为1:(2~15),最优选为1:(3~7)。
在本发明中,上述反应结束后,优选将得到的反应产物透析并冻干,得到式i结构的苯硼酸功能化两性离子嵌段共聚物。本发明对所述透析方式并无限制,采用本领域技术人员熟知的透析方式即可。本发明对所述冻干方式并无限制,采用本领域技术人员熟知的冻干方式即可。
在本发明中,具有式ii结构的聚合物的制备方法优选包括以下步骤:
将具有式iii结构的化合物在具有式iv结构的聚合物的作用下发生开环聚合反应,将得到的反应产物进行脱保护,得到具有式ii结构的聚合物;
式iv中,r1为h或ch3,
r2为磷酰胆碱基团、羧酸甜菜碱基团或磺酸甜菜碱基团。
在本发明中,具有式ii结构的聚合物的制备方法更优选为:
将具有式iii结构的化合物溶解于溶剂中,加入具有式iv结构的聚合物进行开环聚合反应,得到苯甲醇保护的聚合物;
将苯甲醇保护的聚合物进行脱保护,得到具有式ii结构的聚合物。
在本发明中,所述溶剂优选为无水溶剂,更优选为无水n,n-二甲基甲酰胺。在本发明中,式iii结构的化合物和式iv结构的化合物的摩尔比优选为(20~500):1,更优选为(30~300):1,最优选为(40~100):1。
在本发明中,所述开环聚合反应优选在搅拌的条件下进行;所述开环聚合反应的温度优选为20℃~30℃,更优选为23℃~27℃;所述开环聚合反应的时间优选为50h~100h,更优选为60h~80h。
在本发明中,所述开环聚合反应结束后,优选将得到的反应产物沉降、过滤、洗涤、干燥,得到苯甲醇保护的聚合物。在本发明中,所述沉降的试剂优选为乙醚;所述乙醚的体积优选为溶剂的9~11倍体积。本发明对于所述过滤和洗涤方式并无限制,采用本领域技术人员熟知的过滤和洗涤方式即可。在本发明中,所述干燥温度优选为20℃~30℃;所述干燥方式优选为真空干燥。
在本发明中,所述脱保护过程采用的脱保护剂优选为体积浓度为33%(v/v)的氢溴酸。在本发明中,所述脱保护的方法优选为:
将苯甲醇保护的聚合物用二氯乙酸溶解,加入氢溴酸进行脱保护反应。
在本发明中,所述苯甲醇保护的聚合物和氢溴酸的用量比例(质量与体积比例,m/v)优选为1:(1~10),更优选为1:(2~7),最优选为1:(3~5)。
在本发明中,所述脱保护反应温度优选为20℃~30℃,更优选为22℃~28℃;所述脱保护反应的时间优选为0.5h~2h,更优选为1h~1.5h。
在本发明中,反应脱保护反应结束后,优选将得到的反应产物沉降、过滤、溶解、透析并冻干后,得到具有式ii结构的聚合物。
在本发明中,所述式iv结构的聚合物的数均分子量优选为4400~158000,更优选为21700~63300,最优选为28300~47500。
在本发明中,具有式iv结构的聚合物的制备方法优选包括以下步骤:
将式v结构的引发剂引发具有式vi结构的两性离子单体聚合,将得到的聚合物产物脱除boc保护基,得到式iv结构的聚合物。
式vi中,r1为h或ch3,
r2为磷酰胆碱基团、羧酸甜菜碱基团或磺酸甜菜碱基团。
在本发明中,式v中未给出的端基均为甲基。
在本发明中,所述式iv结构的聚合物的制备方法优选为:
将式v结构的引发剂t-boc氨基乙基2溴代异丁酸酯引发式vi结构的两性离子单体聚合并脱除boc保护基制备得到。
本发明优选将式vi结构的两性离子单体溶解于溶剂中,加入式v结构的引发剂t-boc氨基乙基2溴代异丁酸酯进行反应,得到反应产物。
在本发明中,所述溶剂优选为混合溶剂,更优选为甲醇与水的混合溶剂。
在本发明中,所述式vi结构的两性离子单体和式v结构的引发剂的摩尔比优选为(20~500):1,更优选为(100~200):1,最优选为(110~150):1。
在本发明中,所述反应优选在氮气保护的条件下进行。在本发明中,所述反应温度优选为20℃~50℃,更优选为25℃~40℃;所述反应的时间优选为24h~60h,更优选为45h~50h。
在本发明中,所述反应优选在催化剂的条件下进行;所述催化剂优选为cubr/bpy(2,2-联吡啶)体系。在本发明中,所述cubr和bpy的摩尔比优选为1:1~1:5,更优选为1:2。
在本发明中,所述反应结束后,优选将得到的反应溶液暴露于空气中终止反应。本发明优选将反应溶液用甲醇作为洗提液过硅胶柱除去铜盐和bpy,旋转浓缩滤液,经沉降、过滤、洗涤、干燥后,得到boc保护的两性离子聚合物。在本发明中,所述沉降剂优选为thf(四氢呋喃)和乙醚。本发明对所述过滤和洗涤方式并无限制,本领域技术人员熟知的过滤和洗涤方式即可。在本发明中,所述干燥温度优选为20℃~30℃;所述干燥方式优选为真空干燥。
在本发明中,优选将boc保护的两性离子聚合物进行脱保护,得到式iv结构的端氨基两性离子聚合物。在本发明中,所述脱保护采用的脱保护剂优选为三氟乙酸和二氯甲烷的混合溶剂。在本发明中,所述混合溶剂中三氟乙酸的体积浓度(三氟乙酸体积占混合溶剂总体积的百分数)优选为20~30%,更优选为25%。
在本发明中,所述脱保护的方法优选为:
将boc保护的两性离子聚合物(上述反应后得到的产物)用n,n-二甲基甲酰胺溶解,加入脱保护剂进行脱保护反应。
在本发明中,所述boc保护的两性离子聚合物和脱保护剂的摩尔比优选为1:(1~10),更优选为1:(2~7),最优选为1:(3~5)。
在本发明中,所述脱保护反应温度优选为20℃~30℃,更优选为22℃~28℃;所述脱保护反应的时间优选为0.2h~2h,更优选为0.5h~1h。
在本发明中,所述脱保护反应结束后,优选将得到的反应产物沉降、过滤、溶解,调节其ph值。在本发明中,优选为用碳酸氢钠调节ph值,所述ph值优选调制为8~9。在本发明中,调节ph值后,优选为用溶剂萃取,所述萃取溶剂优选为二氯甲烷;本发明对萃取次数并无限制。在本发明中,萃取后,优选将得到的有机相进行干燥、过滤、浓缩得到浓缩产物。在本发明中,所述干燥方式优选为用无水硫酸钠干燥;经过本领域技术人员熟知的过滤和浓缩后,得到浓缩产物;将浓缩产物沉降、过滤、洗涤干燥后得到式iv结构的端氨基两性离子聚合物。在本发明中,所述沉降方式优选为在乙醚中沉降,本发明对于所述过滤、洗涤并无限制,本领域技术人员熟知的过滤、洗涤即可。在本发明中,所述干燥温度优选为20℃~30℃;所述干燥时间优选为20h~30h,更优选为22h~26h;所述干燥方式优选为真空干燥。
本发明提供了一种葡萄糖敏感仿生纳米载体,包括上述技术方案所述的苯硼酸功能化两性离子嵌段共聚物。在本发明中,葡萄糖敏感仿生纳米载体优选为以两性离子聚合物为外壳、以聚谷氨酸酯为内核的纳米胶束。
在本发明中,所述葡萄糖敏感仿生纳米载体优选还包括药物。在本发明中,所述药物优选为胰岛素。在本发明中,所述药物与式i结构的苯硼酸功能化两性离子嵌段共聚物的质量比优选为1:2~9,更优选为1:3~8,最优选为1:4~7。
本发明对所述药物和式i结构的苯硼酸功能化两性离子嵌段共聚物的组合方式并无限制,优选将所述药物包裹在具有式i结构的苯硼酸功能化两性离子嵌段共聚物中。
在本发明中,所述葡萄糖敏感仿生纳米载体的流体力学半径优选为30nm~200nm,更优选为50~150nm,最优选为80~120nm。
本发明提供了一种上述技术方案所述的葡萄糖敏感仿生纳米载体的制备方法,包括:
将上述技术方案所述的苯硼酸功能化两性离子嵌段共聚物在水溶液中自组装,得到葡萄糖敏感仿生纳米载体。
在本发明中,两性离子仿生聚合物为亲水段形成胶束的外壳,赋予纳米载体良好的生物相容性和血液循环时间,聚谷氨酸酯形成疏水的内核并赋予纳米载体良好的葡萄糖敏感性能。
在本发明中,所述葡萄糖敏感仿生纳米载体的制备方法优选为:
将式i结构的苯硼酸功能化两性离子嵌段共聚物溶于溶剂中,并加入去离子水,然后透析得到葡萄糖敏感仿生纳米载体。
在本发明中,所述溶剂优选为有机溶剂,更优选为n,n-二甲基甲酰胺或二甲基亚砜。本发明对于所述透析方式并无限制,本领域技术人员熟知的透析方式即可。
在本发明中,所述葡萄糖敏感仿生纳米载体的制备方法更优选为:
将式i结构的苯硼酸功能化两性离子嵌段共聚物溶解于溶剂中,得到第一溶液;
向第一溶液加入药物的水溶液,透析后得到葡萄糖敏感仿生纳米载体。
在本发明中,所述药物与式i结构的苯硼酸功能化两性离子嵌段共聚物的质量比优选为1:2~9,更优选为1:3~8,最优选为1:4~6。
在本发明中,所述药物优选为胰岛素,所述胰岛素在葡萄糖敏感仿生纳米载体中的理论载药量优选为5%~30%,更优选为10~25%,最优选为15~25%。在本发明中,所述溶剂优选为磷酸盐缓冲液。在本发明中,所述加入药物水溶液优选为滴加的方式加入。
在本发明中,加入药物水溶液后,优选为搅拌并透析得到葡萄糖敏感仿生纳米药物载体;所述搅拌的时间优选为12~24h;所述透析优选为用缓冲溶液透析后用去离子水透析。
本发明提供的葡萄糖敏感仿生纳米药物载体由式i结构的苯硼酸功能化两性离子嵌段共聚物自组装得到,这种葡萄糖敏感仿生纳米载体在水中具有亲水链段向外、疏水链段向内的纳米胶束形态,药物可被包裹于葡萄糖敏感仿生纳米载体中,具体的说,药物可被包裹在葡萄糖敏感仿生纳米载体的疏水内核中,从而实现缓释。
本发明中,葡萄糖敏感仿生纳米载体中,式i结构的苯硼酸功能化两性离子嵌段共聚物具有亲水性、聚氨基酸链段具有疏水性能够担载药物,且经由苯硼酸修饰的葡萄糖敏感仿生纳米载体在葡萄糖的存在下,小分子的葡萄糖与式i结构中的苯硼酸形成硼酯键,致使葡萄糖敏感仿生纳米载体体积变大,亲水性增加,进而将担载的药物快速释放出来,因而具有较强的葡萄糖敏感性。
与现有技术相比,本发明提供了具有式i结构的苯硼酸功能化两性离子嵌段共聚物,这种共聚物包括两性离子仿生聚合物亲水链段和聚谷氨酸酯疏水链段,其中,两性离子仿生聚合物和聚谷氨酸酯均具有良好的生物相容性,聚谷氨酸酯具有良好的生物降解性,且聚谷氨酸酯经由苯硼酸修饰,具有葡萄糖敏感性能,苯硼酸功能化两性离子嵌段共聚物具有潜在的体内长循环性能。因此,采用苯硼酸功能化两性离子嵌段共聚物制备的葡萄糖敏感仿生纳米载体具有良好的生物相容性和生物降解性,能在生物体内降解而不会对生物体产生危害;因苯硼酸的存在,该载体可对葡萄糖浓度的变化作出迅速的反应,有利于药物的迅速释放,提高药物的疗效;苯硼酸功能化两性离子嵌段共聚物的存在,具有潜在的纳米药物载体体内长循环特性,有望进一步提高疗效。
综上,本发明提供了由式i结构的仿生嵌段共聚物即苯硼酸功能化两性离子嵌段共聚物得到的葡萄糖敏感仿生纳米载体,葡萄糖敏感仿生纳米载体由包括两性离子仿生聚合物亲水链段和苯硼酸修饰的聚谷氨酸酯疏水链段的聚合物,其中,两性离子仿生聚合物和聚谷氨酸酯均具有良好的生物相容性,聚谷氨酸酯具有良好的生物降解性,因此,本发明中的葡萄糖敏感仿生纳米载体具有良好的生物相容性和生物降解性,能在生物体内降解而不会对生物体产生危害;葡萄糖敏感仿生纳米载体中的苯硼酸基团具有葡萄糖敏感性,可对葡萄糖浓度的变化作出迅速的反应,有利于药物的迅速释放,提高药物的疗效;两性离子仿生聚合物能够延长纳米载体的血液循环时间,有望增强纳米药物载体的疗效。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
图1为本发明实施例13制备的葡萄糖敏感仿生纳米载体与葡萄糖浓度的曲线图;
图2为本发明实施例14提供的葡萄糖敏感仿生纳米载体的累计释放百分比与葡萄糖浓度的曲线图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
在配有磁子的10ml单口烧瓶中,加入式v结构的引发剂、羧基甜菜碱丙烯酸甲酯(cbma)单体(9.17g,40mmo1)和90mlh2o/ch3oh(1:l,v/v)混合溶剂。搅拌充分溶解后,在氮气保护下经过三次冷冻、抽气、融化循环,再加入已除氧的cubr(7.2mg,0.05mmo1)、bpy(16mg,0.1mmo1),在35℃下反应48h。反应结束后,将溶液暴露于空气中终止反应。将得到的反应溶液用甲醇作为洗提液过硅胶柱除去铜盐和bpy。旋转浓缩滤液,乙醚沉降、过滤,真空干燥得到boc保护的两性离子聚合物。
将该boc保护的两性离子聚合物溶于50mldmf中,并滴加16ml三氟乙酸溶液(三氟乙酸/二氯甲烷:25%,v/v)溶液25℃下反应40min。反应结束后,将反应溶液用乙醚沉降、过滤,用碳酸氢钠水溶液洗涤,用二氯甲烷萃取,并用无水硫酸钠干燥后,过滤、浓缩、沉降、真空干燥得到端氨基两性离子聚合物pcbma-nh2;
对聚合物pcbma-nh2进行核磁共振检测,检测结果为cbma的聚合度是50,其结构为:
其分子量为9380。
实施例2
在配有磁子的10ml单口烧瓶中,加入式v结构的引发剂(310.18mg,1mmol)、硫代甜菜碱丙烯酸甲酯(sbma)单体(22.35g,80mmo1)和200mlh2o/ch3oh(1:l,v/v)混合溶剂。搅拌充分溶解后,在氮气保护下经过三次冷冻、抽气、融化循环,再加入已除氧的cubr(7.2mg,0.05mmo1)、bpy(16mg,0.1mmo1),在35℃下反应48h。反应结束后,将反应溶液暴露于空气中终止反应。将反应溶液用甲醇作为洗提液过硅胶柱除去铜盐和bpy。旋转浓缩滤液,乙醚沉降、过滤,真空干燥得到boc保护的两性离子聚合物。
将该boc保护的两性离子聚合物溶于100mldmf中,并滴加16ml三氟乙酸溶液(三氟乙酸/二氯甲烷:25%,v/v)溶液25℃下反应40min。反应结束后,将反应溶液用乙醚沉降、过滤,用碳酸氢钠水溶液洗涤,用二氯甲烷萃取,并用无水硫酸钠干燥后,过滤、浓缩、沉降、真空干燥得到端氨基两性离子聚合物psbma-nh2。
对聚合物psbma-nh2进行核磁共振检测,检测结果为sbma的聚合度是80,其结构为:
所得产物分子量为22500。
实施例3
在配有磁子的10ml单口烧瓶中,加入式v结构的引发剂(310.18mg,1mmol)、甲基丙烯酸磷酸胆碱(mpc)单体(33g,110mmo1)和300mlh2o/ch3oh(1:l,v/v)混合溶剂。搅拌充分溶解后,在氮气保护下经过三次冷冻、抽气、融化循环,再加入已除氧的cubr(7.2mg,0.05mmo1)、bpy(16mg,0.1mmo1),在35℃下反应48h。反应结束后,将溶液暴露于空气中终止反应。将反应溶液用甲醇作为洗提液过硅胶柱除去铜盐和bpy。旋转浓缩滤液,乙醚沉降、过滤,真空干燥得到boc保护的两性离子聚合物。
将该boc保护的两性离子聚合物溶于150mldmf中,并滴加16ml三氟乙酸溶液(三氟乙酸/二氯甲烷:25%,v/v)溶液25℃下反应40min。反应结束后,将反应溶液用乙醚沉降、过滤,用碳酸氢钠水溶液洗涤,用二氯甲烷萃取,并用无水硫酸钠干燥后,过滤、浓缩、沉降、真空干燥得到端氨基两性离子聚合物pmpc-nh2。
对聚合物pmpc-nh2进行核磁共振检测,检测结果证实甲基丙烯酸磷酸胆碱的聚合度为110,,其结构为:
所得产物分子量为33100。
实施例4
在无水条件下向反应瓶中加入1.951g(0.208mmol)实施例1制备的端氨基两性离子聚合物pcbma-nh2,采用甲苯共沸除水后用无水n,n-二甲基甲酰胺溶解,得到端氨基两性离子聚合物pcbma-nh2溶液。
将3.2854g(12.48mmol)式iii结构的化合物用无水n,n-二甲基甲酰胺溶解,并加入到所述端氨基两性离子聚合物pcbma-nh2溶液中,在25℃、搅拌子搅拌条件下进行反应,反应时间为72h,反应结束后,将反应溶液倒入体积为溶剂10倍量的乙醚中沉降、过滤、洗涤、25℃真空干燥24h,得到苯甲醇保护的两性离子仿生嵌段共聚物(pcbma-b-pblg)。
将苯甲醇保护的两性离子仿生嵌段共聚物(pcbma-b-pblg)溶解于34ml二氯乙酸中,然后在室温下加入10.2ml质量浓度为33%的溴化氢的冰醋酸溶液,在30℃下搅拌60min后,将得到的产物用乙醚沉降并用乙醚洗涤;然后将产物溶于n,n-二甲基甲酰胺中,用3500da的透析袋透析3天,冻干后得到两性离子仿生嵌段共聚物(pcbma50-b-pga60)。
将制备得到的两性离子仿生嵌段共聚物(pcbma50-b-pga60)用1hnmr测定其数均分子量,并计算其平均聚合度,以及计算该反应的反应产率,结果见表1,其结构式为:
实施例5
在无水条件下向反应瓶中加入0.976g(0.104mmol)实施例1制备的端氨基两性离子聚合物pcbma-nh2,采用甲苯共沸除水后用无水n,n-二甲基甲酰胺溶解,得到端氨基两性离子聚合物pcbma-nh2溶液。
将3.2854g(12.48mmol)式iii结构的化合物用无水n,n-二甲基甲酰胺溶解,并加入到所述端氨基两性离子聚合物pcbma-nh2溶液中,在25℃、搅拌子搅拌条件下进行反应,反应时间为72h,反应结束后,将反应溶液倒入体积为溶剂10倍量的乙醚中沉降、过滤、洗涤、25℃真空干燥24h,得到苯甲醇保护的两性离子仿生嵌段共聚物(pcbma-b-pblg)。
将制备的苯甲醇保护的两性离子仿生嵌段共聚物(pcbma-b-pblg)溶解于31ml二氯乙酸中,然后在室温下加入10.2ml质量浓度为33%的溴化氢的冰醋酸溶液,在30℃下搅拌60min后,将得到的产物用乙醚沉降并用乙醚洗涤;然后将产物溶于n,n-二甲基甲酰胺中,用3500da的透析袋透析3天,冻干后得到两性离子仿生嵌段共聚物(pcbma50-b-pga120)。
将上述制备两性离子仿生嵌段共聚物(pcbma50-b-pga120)用1hnmr测定其数均分子量,并计算其平均聚合度,以及计算该反应的反应产率,结果见表1。
实施例6
在无水条件下向反应瓶中加入4.680g(0.208mmol)实施例1制备的端氨基两性离子聚合物psbma-nh2,采用甲苯共沸除水后用无水n,n-二甲基甲酰胺溶解,得到端氨基两性离子聚合物pcbma-nh2溶液。
将3.2854g(12.48mmol)式iii结构的化合物用无水n,n-二甲基甲酰胺溶解,并加入到所述端氨基两性离子聚合物pcbma-nh2溶液中,在25℃、搅拌子搅拌条件下进行反应,反应时间为72h,反应结束后,将反应溶液倒入体积为溶剂10倍量的乙醚中沉降、过滤、洗涤、25℃真空干燥24h,得到苯甲醇保护的两性离子仿生嵌段共聚物(psbma-b-pblg)。
将苯甲醇保护的两性离子仿生嵌段共聚物(psbma-b-pblg)溶解于34ml二氯乙酸中,然后在室温下加入10.2ml质量浓度为33%的溴化氢的冰醋酸溶液,在30℃下搅拌60min后,将得到的产物用乙醚沉降并用乙醚洗涤;然后将产物溶于n,n-二甲基甲酰胺中,用3500da的透析袋透析3天,冻干后得到两性离子仿生嵌段共聚物(psbma80-b-pga60)。
将制备得到的两性离子仿生嵌段共聚物(psbma80-b-pga60)用1hnmr测定其数均分子量,并计算其平均聚合度,以及计算该反应的反应产率,结果见表1。
实施例7
在无水条件下向反应瓶中加入2.340g(0.104mmol)实施例1制备的端氨基两性离子聚合物psbma-nh2,采用甲苯共沸除水后用无水n,n-二甲基甲酰胺溶解,得到端氨基两性离子聚合物pcbma-nh2溶液。
将3.2854g(12.48mmol)式iii结构的化合物用无水n,n-二甲基甲酰胺溶解,并加入到所述端氨基两性离子聚合物pcbma-nh2溶液中,在25℃、搅拌子搅拌条件下进行反应,反应时间为72h,反应结束后,将反应溶液倒入体积为溶剂10倍量的乙醚中沉降、过滤、洗涤、25℃真空干燥24h,得到苯甲醇保护的两性离子仿生嵌段共聚物(psbma-b-pblg)。
将苯甲醇保护的两性离子仿生嵌段共聚物(psbma-b-pblg)溶解于60ml二氯乙酸中,然后在室温下加入10.2ml质量浓度为33%的溴化氢的冰醋酸溶液,在30℃下搅拌60min后,将得到的产物用乙醚沉降并用乙醚洗涤;然后将产物溶于n,n-二甲基甲酰胺中,用3500da的透析袋透析3天,冻干后得到两性离子仿生嵌段共聚物(psbma80-b-pga120)。
将制备得到的两性离子仿生嵌段共聚物(psbma80-b-pga120)用1hnmr测定其数均分子量,并计算其平均聚合度,以及计算该反应的反应产率,结果见表1。
实施例8
在无水条件下向反应瓶中加入6.885g(0.208mmol)实施例1制备的端氨基两性离子聚合物pmpc-nh2,采用甲苯共沸除水后用无水n,n-二甲基甲酰胺溶解,得到端氨基两性离子聚合物pcbma-nh2溶液。
将3.2854g(12.48mmol)式iii结构的化合物用无水n,n-二甲基甲酰胺溶解,并加入到所述端氨基两性离子聚合物pcbma-nh2溶液中,在25℃、搅拌子搅拌条件下进行反应,反应时间为72h,反应结束后,把溶液倒入体积为溶剂10倍量的乙醚中沉降、过滤、洗涤、25℃真空干燥24h,得到苯甲醇保护的两性离子仿生嵌段共聚物(pmpc-b-pblg)。
将苯甲醇保护的两性离子仿生嵌段共聚物(pmpc-b-pblg)溶解于100ml二氯乙酸中,然后在室温下加入10.2ml质量浓度为33%的溴化氢的冰醋酸溶液,在30℃下搅拌60min后,将得到的产物用乙醚沉降并用乙醚洗涤;然后将产物溶于n,n-二甲基甲酰胺中,用3500da的透析袋透析3天,冻干后得到两性离子仿生嵌段共聚物(pmpc110-b-pga60)。
将制备得到的两性离子仿生嵌段共聚物(pmpc110-b-pga60)用1hnmr测定其数均分子量,并计算其平均聚合度,以及计算该反应的反应产率,结果见表1。
实施例9
在无水条件下向反应瓶中加入3.442g(0.104mmol)实施例1制备的端氨基两性离子聚合物pmpc-nh2,采用甲苯共沸除水后用无水n,n-二甲基甲酰胺溶解,得到端氨基两性离子聚合物pcbma-nh2溶液。
将3.2854g(12.48mmol)式iii结构的化合物用无水n,n-二甲基甲酰胺溶解,并加入到所述端氨基两性离子聚合物pcbma-nh2溶液中,在25℃、搅拌子搅拌条件下进行反应,反应时间为72h,反应结束后,将反应溶液倒入体积为溶剂10倍量的乙醚中沉降、过滤、洗涤、25℃真空干燥24h,得到苯甲醇保护的两性离子仿生嵌段共聚物(pmpc-b-pblg)。
将苯甲醇保护的两性离子仿生嵌段共聚物(pmpc-b-pblg)溶解于70ml二氯乙酸中,然后在室温下加入10.2ml质量浓度为33%的溴化氢的冰醋酸溶液,在30℃下搅拌60min后,将得到的产物用乙醚沉降并用乙醚洗涤;然后将产物溶于n,n-二甲基甲酰胺中,用3500da的透析袋透析3天,冻干后得到两性离子仿生嵌段共聚物(pmpc110-b-pga120)。
将制备得到的两性离子仿生嵌段共聚物(pmpc110-b-pga120)用1hnmr测定其数均分子量,并计算其平均聚合度,以及计算该反应的反应产率,结果见表1。
表1本发明实施例4~9制备的两性离子仿生嵌段共聚物的性能参数
表1中,m/i为式iii结构的化合物与端氨基两性离子聚合物的摩尔数的比值;mn为两性离子仿生嵌段共聚物的数均分子量;dp为两性离子仿生嵌段共聚物中谷氨酸的平均聚合度;反应产率为实际得到的两性离子仿生嵌段共聚物的质量与理论得到的两性离子仿生嵌段共聚物质量的比值。
实施例10
将实施例6制备的两性离子仿生嵌段共聚物1.495g(数均分子量为29900的psbma80-b-pga570.05mmol)在25℃下用n,n-二甲基甲酰胺溶解,然后在搅拌子搅拌下加入2.473g(12.90mmol)1-(3-二甲基氨基丙基)-3-乙基碳化二亚胺盐酸盐(edc.hcl)和0.983g(8.55mmol)n-羟基琥珀酰亚胺(nhs)进行活化反应过夜。向活化好的反应体系中加入0.1394g(0.90mmol)3-氨基苯硼酸一水合物进行缩合反应,反应结束后经过透析、冷冻干燥得到苯硼酸功能化两性离子嵌段共聚物。
对制备得到的苯硼酸功能化两性离子嵌段共聚物进行核磁共振检测,检测结果参见表2,其结构式为:
实施例11
将实施例6制备的两性离子仿生嵌段共聚物(psbma80-b-pga57)1.495g数均分子量为29900的psbma80-b-pga57(0.05mmol),在25℃下用n,n-二甲基甲酰胺溶解,然后在搅拌子搅拌下加入2.473g(12.90mmol)1-(3-二甲基氨基丙基)-3-乙基碳化二亚胺盐酸盐(edc.hcl)和0.983g(8.55mmol)n-羟基琥珀酰亚胺(nhs)进行活化反应过夜。向活化好的反应体系中加入0.2633g(1.70mmol)3-氨基苯硼酸一水合物进行缩合反应,反应结束后经过透析、冷冻干燥得到苯硼酸功能化两性离子嵌段共聚物。
对制备得到的苯硼酸功能化两性离子嵌段共聚物进行核磁共振检测,检测结果参见表2。
实施例12
将实施例6制备的两性离子仿生嵌段共聚物(psbma80-b-pga57)1.495g数均分子量为29900的psbma80-b-pga57(0.05mmol)在25℃下用n,n-二甲基甲酰胺溶解,然后在搅拌子搅拌下加入2.473g(12.90mmol)1-(3-二甲基氨基丙基)-3-乙基碳化二亚胺盐酸盐(edc.hcl)和0.983g(8.55mmol)n-羟基琥珀酰亚胺(nhs)进行活化反应过夜。向活化好的反应体系中加入0.3874g(2.5mmol)3-氨基苯硼酸一水合物进行缩合反应,反应结束后经过透析、冷冻干燥得到苯硼酸功能化两性离子嵌段共聚物。
对制备得到的苯硼酸功能化两性离子嵌段共聚物进行核磁共振检测,检测结果参见表2。
表2本发明实施例10~12制备的苯硼酸功能化两性离子嵌段共聚物性能参数
实施例13
将实施例11制备的苯硼酸功能化两性离子嵌段共聚物0.341g(mn=34100)溶解于10ml二甲基亚砜中(含少量碳酸氢钠),将去离子水滴加到二甲基亚砜中,搅拌过夜后透析得到的葡萄糖敏感仿生纳米载体溶液,将葡萄糖敏感仿生纳米载体浓度定为0.2mg/ml。
取5.0ml、浓度为0.2mg/ml的上述葡萄糖敏感仿生纳米载体溶液,其中含葡萄糖浓度分别为0、0.5、1.0、1.5、2.0、2.5、3.0、4.0和5.0mg/ml。
采用动态光散射(dls)方法分别检测上述葡萄糖敏感仿生纳米载体的粒径,检测结果参见图1,图1为本发明实施例13制备的葡萄糖敏感仿生纳米载体与葡萄糖浓度的曲线图。由图1可知,葡萄糖敏感仿生纳米载体随着葡萄糖浓度的增加而变大;在2.0~3.0mg/ml葡萄糖浓度溶液中葡萄糖敏感仿生纳米载体的粒径变化较大;当葡萄糖浓度大于3.0mg/ml时,葡萄糖敏感仿生纳米载体粒径的变化趋势减小,葡萄糖浓度为5.0mg/ml时粒径几乎达到最大值;由此可知,该葡萄糖敏感仿生纳米载体在较低葡萄糖浓度时即具有良好的葡萄糖敏感性。
实施例14
将实施例11制备的苯硼酸功能化两性离子嵌段共聚物0.341g(mn=34100)溶解于10ml二甲基亚砜中(含少量碳酸氢钠),将含胰岛素0.068g的pbs溶液滴加到二甲基亚砜中,搅拌过夜后透析并冷冻干燥得到的葡萄糖敏感仿生纳米载体。
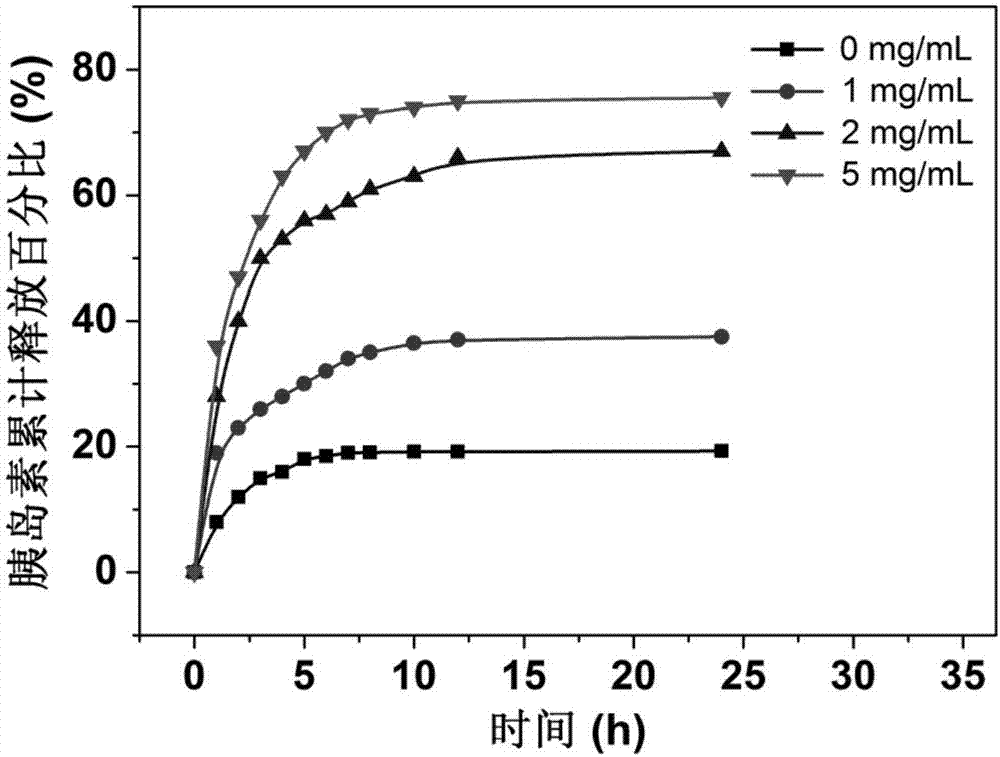
称取上述葡萄糖敏感仿生纳米载体5mg用3ml的pbs溶液溶解后置于截留分子量为7000的透析袋中,并将该透析袋放入含15mlpbs溶液的小烧杯中进行药物体外释放实验。控制pbs溶液中葡萄糖浓度为0、1.0、2.0和5.0mg/ml。将小烧杯置于37.5℃振荡箱中,定时取出1ml释放液,并补充相应新鲜缓冲液。采用bca法测定释放液中胰岛素的浓度并采用累积计算的方法计算胰岛素的累积释放百分比。检测结果参见图2,图2为本发明实施例14提供的葡萄糖敏感仿生纳米载体的累计释放百分比与葡萄糖浓度的曲线图。
由图2可知,胰岛素的累计释放百分比随着葡萄糖浓度的增加而变大;在同一葡萄糖浓度下,随着时间的延长胰岛素的累积释放量增加。胰岛素的释放速率在2.0mg/ml葡萄糖溶液中变化较大;当葡萄糖浓度为1.0mg/ml时,胰岛素的释放速率较小,表明该载药纳米胶束能够在糖尿病患者血糖浓度下快速大量的释放胰岛素,而在正常血糖水平较少释放胰岛素,具有较好的葡萄糖敏感药物释放特性。
由以上实施例可知,本发明提供了一种苯硼酸功能化两性离子嵌段共聚物,具有式i结构。本发明提供了一种葡萄糖敏感仿生纳米载体,由苯硼酸功能化两性离子嵌段共聚物在水溶液中自组装制备得到,苯硼酸功能化两性离子嵌段共聚物和聚谷氨酸酯均具有良好的生物相容性,聚谷氨酸酯具有良好的生物降解性,且聚谷氨酸酯经由苯硼酸修饰。本发明提供的葡萄糖敏感仿生纳米载体具有良好的生物相容性和生物降解性;因苯硼酸与1,2-二元醇的特殊成硼酯键反应赋予载体具有葡萄糖敏感性能,可对葡萄糖浓度变化作出迅速反应,有利于药物迅速释放,提高药物疗效。苯硼酸功能化两性离子嵌段共聚物具有体内长循环特性,有望进一步提高疗效。
- 还没有人留言评论。精彩留言会获得点赞!