甲磺酸达比加群酯的晶型VII及其制备方法与流程
本发明属于药物结晶技术领域,特别涉及一种新的甲磺酸达比加群酯晶型及其结晶制备方法。
背景技术:
甲磺酸达比加群酯的中文化学名为3-[[[2-[[[4-[[[(己氧基)羰基]氨基]亚氨甲基]苯基]氨基]甲基]-1-甲基-1h-苯并咪唑-5-基]羰基](吡啶-2-基)氨基]丙酸乙酯甲磺酸盐,英文化学名为ethyl3-[[[2-[[[4-[[[(hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1h-benzimidazol-5-yl]carbonyl](pyridin-2-yl)amino]propionatemethanesulfonate,分子式为c34h41n7o5·ch4o3s,分子量为723.86。其结构式如式(i)所示。
达比加群(dabigatran)最早公开于wo98/37075中,后由德国勃林格殷格翰公司(boehringeringelheim)研制开发,于2008年4月首次在德国和英国上市,商品名pradaxa,药用成分为甲磺酸达比加群酯。该药是继华法林之后50年来首个上市的全新口服直接抗凝血药物,2008年在欧盟获准用于全髋或全膝关节置换手术术后静脉血栓的预防。该药口服后在体内释放出达比加群,与凝血酶的纤维蛋白特异位点结合,阻止纤维蛋白原裂解为纤维蛋白,从而阻断凝血瀑布网络的最后步骤及血栓形成。2010年10月,fda又批准甲磺酸达比加群酯胶囊用于非瓣膜性房颤患者预防血栓和脑卒中的发生。甲磺酸达比加群酯不但具有有效、可预测且一致的抗凝作用以及良好的卒中预防作用,而且具有出血风险较低,无需常规监测等优点,药物间相互作用的可能性较低,且不与食物发生相互作用。随着该药在中国问世,达比加群酯将为我国近千万心房颤动患者在预防卒中和全身性栓塞方面,提供新的治疗手段。
活性药物成分除了在稳定性、吸湿性、可溶性需要满足一定的要求之外,其结晶变体形态也应该有严格的控制。
达比加群酯与多种无机酸和有机酸形成的加成盐有多种晶型。专利wo2008043759a1和wo2011110876a1公开了2,5-二羟基苯甲酸盐、苯磺酸盐、盐酸盐、富马酸盐、草酸盐、磷酸盐、水杨酸盐等盐的不同晶型的制备方法。
甲磺酸达比加群酯的晶体有多种晶型,专利文献的报道中有如下晶型和无定型:
专利zl200480024952.1公开了甲磺酸达比加群酯的晶型i、晶型ii以及半水合物;
专利zl201110249228.0公开了甲磺酸达比加群酯的一水合物;
专利wo2012027543a1公开了甲磺酸达比加群酯晶型a、b、c、d、g、h、iii以及无定形;
专利wo2011110876a1公开了甲磺酸达比加群酯晶型iv。
专利cn103951654a公开了甲磺酸达比加群酯晶型v。
专利cn103965164a公开了甲磺酸达比加群酯晶型vi。
尽管已有上述多种晶型的报告,但它们普遍存在粒度较小,堆密度小,流动性差,存在安全隐患等问题,因此仍有必要研究环境污染小,方便易得、热力学稳定性好,便于工业化生产的甲磺酸达比加群酯新晶型,以保证原料药及其制剂在制备和存储中的稳定性,提高甲磺酸达比加群酯的药物质量和临床疗效。
技术实现要素:
本发明公开一种甲磺酸达比加群酯新晶型及其结晶制备方法。
本发明公开的甲磺酸达比加群酯的新晶型,其x-射线粉末衍射在衍射角2θ为2.8±0.2,5.7±0.2,10.6±0.2,11.5±0.2,11.9±0.2,17.3±0.2,17.8±0.2,18.2±0.2,19.8±0.2,21.3±0.2,24.1±0.2°处有特征峰,称为vii晶型。
在一种优选方案中,本vii晶型的x-射线粉末衍射在衍射角2θ为2.8±0.2,5.7±0.2,10.6±0.2,11.5±0.2,11.9±0.2,12.2±0.2,12.8±0.2,13.9±0.2,14.5±0.2,15.9±0.2,17.3±0.2,17.8±0.2,18.2±0.2,19.8±0.2,21.3±0.2,21.7±0.2,24.1±0.2,26.2±0.2,27.3±0.2,29.3±0.2°处有特征峰,如图1所示。
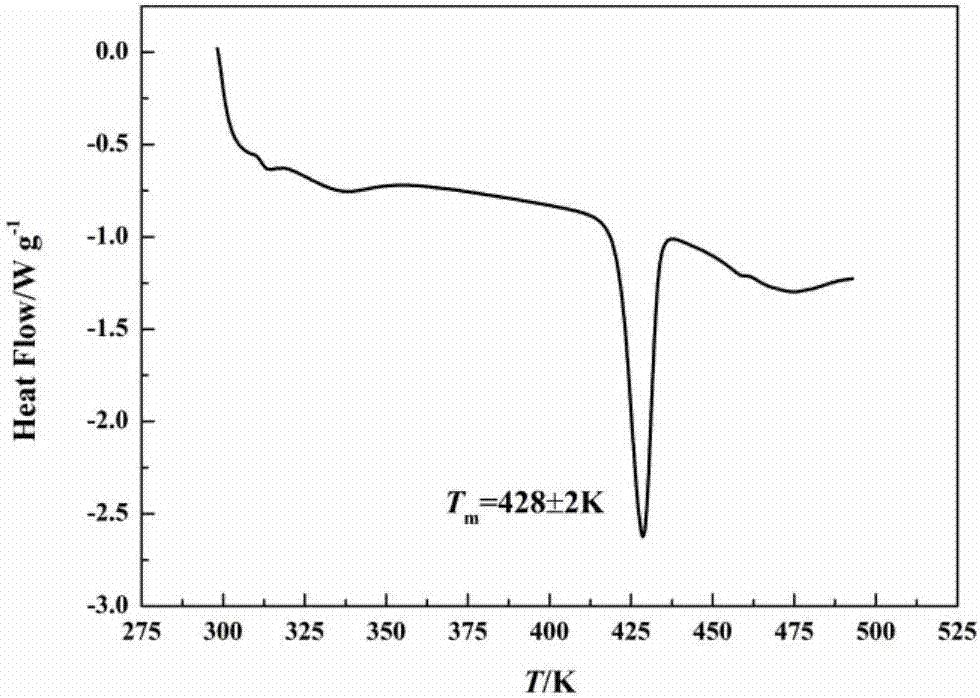
所述vii晶型的dsc分析结果显示,在428±2k处有一个吸热峰,如图2所示。
所述vii晶型的红外光谱在3305±2cm-1、2933±2cm-1、2861±2cm-1、1736±2cm-1、1651±2cm-1、1608±2cm-1、1577±2cm-1、1470±2cm-1、1439±2cm-1、1379±2cm-1、1323±2cm-1、1234±2cm-1、1165±2cm-1、1039±2cm-1、1000±2cm-1、950±2cm-1、831±2cm-1、776±2cm-1、556±2cm-1、519±2cm-1处有吸收峰,具体如图3所示。
所述vii晶型的拉曼光谱在1751±2cm-1、1739±2cm-1、1649±2cm-1、1620±2cm-1、1602±2cm-1、1571±2cm-1、1523±2cm-1、1405±2cm-1、1381±2cm-1、1348±2cm-1、1319±2cm-1、1241±2cm-1、1210±2cm-1、1162±2cm-1、1109±2cm-1、1040±2cm-1、1007±2cm-1、993±2cm-1、953±2cm-1、847±2cm-1、805±2cm-1、774±2cm-1、750±2cm-1、730±2cm-1、663±2cm-1、626±2cm-1、579±2cm-1、555±2cm-1、520±2cm-1、405±2cm-1、353±2cm-1、311±2cm-1、255±2cm-1处有吸收峰,具体如图4所示。
本发明所述的vii晶型的制备方法,具体包括如下步骤:
a)将甲磺酸达比加群酯粗品加入无水乙醇中,配置成0.05~0.13g溶质/g溶剂的悬浮液;
b)15~25℃恒温悬浮搅拌条件下,微波处理5-10min;
c)微波处理后,体系继续在15~25℃恒温条件下悬浮搅拌8-24h,抽滤悬浮液,干燥晶体产品至恒重。
在上述制备方法中,所述的甲磺酸达比加群酯粗品为晶型i固体粉末,hplc纯度≥95%。
在上述制备方法中,微波功率为100-200w。
在上述制备方法中,干燥在35℃-60℃、常压的条件下进行。
在上述制备方法中,结晶方式为微波辅助结晶。
本申请公开的甲磺酸达比加群酯晶型vii的制备方法是在外部微波场的作用下使晶体中分子排列方式及构象产生变化,从而促成了新晶型的生成。在普通的悬浮转晶工艺条件下,晶型vii不能成功制备,说明了外部微波场对新晶型制备的必要性及有效性,这可能与微波产生的局部高温高压等条件密切相关。
对本发明所述产品进行长期稳定性试验,其在常温常压条件下贮存4个月后颜色、形态未发生明显变化,产品稳定性良好,其x-射线粉末衍射及dsc分析考察结果如图5、图6所示。
表1本申请产品和现有技术产品对比
与现有技术相比,本发明提供的甲磺酸达比加群酯晶型vii的制备方法具有所用溶剂毒性低、环境污染小,工艺简单、适于工业化生产等优点。同时本发明提供的方法制备出的甲磺酸达比加群酯晶型vii的溶解度较晶型i提高了约70%,较晶型ii提高了约120%。此外,甲磺酸达比加群酯的晶型i,ii以及半水合物产品的粒度都比较小,因此产品聚结问题非常严重,多为20μm的片状聚结物,过滤及产品后处理都很困难;而新制备的晶型vii则为棒状晶体不规则球形聚结体,表观粒度为200μm左右,大大提高了产品的流动性及过滤性质,晶型i与晶型vii的扫描电镜图如图7、图8所示。因此,晶型vii具有容易过滤、溶解度高、纯度高、稳定性好等优点,有利于后续的制剂、储存和运输,在药物制剂中占有很大的优势。
附图说明
图1:甲磺酸达比加群酯晶型vii的x-射线粉末衍射图;
图2:甲磺酸达比加群酯晶型vii的dsc分析图;
图3:甲磺酸达比加群酯晶型vii的红外光谱图;
图4:甲磺酸达比加群酯晶型vii的拉曼光谱图;
图5:甲磺酸达比加群酯晶型vii的稳定性研究x-射线粉末衍射图;
图6:甲磺酸达比加群酯晶型vii的稳定性研究dsc分析图。
图7:甲磺酸达比加群酯晶型vii的扫描电镜图。
图8:甲磺酸达比加群酯晶型i的扫描电镜图。
具体实施方式
实施例1:
将干燥的甲磺酸达比加群酯晶型i30.20g加入300.04g无水乙醇中形成悬浮液,在搅拌下将该悬浮液恒温于25℃,恒定温度,微波处理5min,功率为100w,微波处理结束后继续悬浮搅拌18h,抽滤悬浮液,将产物在60℃、常压下干燥至恒重,得到vii晶型甲磺酸达比加群酯。
本实施例所得晶体的hplc纯度为99.18%。
本实施例制得的晶体的x-射线粉末衍射如图1所示:其主要在衍射角2θ=2.800,5.702,10.600,11.500,11.878,17.321,17.781,18.180,19.804,21.259,24.080°处有特征峰,更全面的特征峰为衍射角2θ为2θ为2.800,5.702,10.600,11.500,11.878,12.162,12.759,13.872,14.521,15.858,17.321,17.781,18.180,19.804,21.259,21.740,24.080,26.179,27.278,29.318°
本实施例制得的晶体的dsc图谱如图2所示:在428k处有吸热峰。
本实施例制得的晶体的红外图谱如图3所示:在3304.93cm-1、2933.06cm-1、2861.21cm-1、1735.90cm-1、1650.67cm-1、1608.04cm-1、1577.13cm-1、1470.04cm-1、1439.41cm-1、1378.76cm-1、1322.93cm-1、1233.86cm-1、1165.41cm-1、1039.19cm-1、999.82cm-1、949.84cm-1、830.84cm-1、776.24cm-1、556.14cm-1、519.44cm-1等处有吸收峰。
本实施例制得的晶体的拉曼图谱如图4所示:1751.4cm-1、1738.6cm-1、1649.2cm-1、1620.1cm-1、1601.8cm-1、1570.8cm-1、1523.4cm-1、1404.8cm-1、1381.1cm-1、1348.3cm-1、1319.1cm-1、1240.7cm-1、1209.7cm-1、1161.7cm-1、1109.4cm-1、1040.1cm-1、1007.2cm-1、992.6cm-1、952.5cm-1、846.7cm-1、804.8cm-1、773.8cm-1、750.1cm-1、730.0cm-1、662.5cm-1、626.0cm-1、578.6cm-1、554.9cm-1、520.3cm-1、405.3cm-1、352.5cm-1、310.5cm-1、255.0cm-1等处有吸收峰。
本实施例所得晶体产品在给定的测试溶剂中具有更高的溶解度,结果如表1所示。
稳定性研究数据显示,本实施例所得晶体产品稳定性良好,常温密封放置3天、2个月、2.5个月、3个月、4个月后,晶型vi产品晶型均并未发生变化。稳定性图谱参见附图5、附图6。
实施例2:
将干燥的甲磺酸达比加群酯晶型i0.65g加入5.00g无水乙醇中形成悬浮液,在搅拌下将该悬浮液恒温于25℃,恒定温度,微波处理5min,功率为100w,微波处理结束后继续悬浮搅拌8h,抽滤悬浮液,将产物在35℃、常压下干燥至恒重,得到vii晶型甲磺酸达比加群酯。
本实施例所得晶体的hplc纯度为99.08%,表征数据与实施例1相同。
实施例3:
将干燥的甲磺酸达比加群酯晶型i3.02g加入40.22g无水乙醇中形成悬浮液,在搅拌下将该悬浮液恒温于20℃,恒定温度,微波处理10min,功率为150w,微波处理结束后继续悬浮搅拌10h,抽滤悬浮液,将产物在60℃、常压下干燥至恒重,得到vii晶型甲磺酸达比加群酯。
本实施例所得晶体的hplc纯度为99.28%,表征数据与实施例1相同。
实施例4:
将干燥的甲磺酸达比加群酯晶型i0.25g加入5.00g无水乙醇中形成悬浮液,在搅拌下将该悬浮液恒温于15℃,恒定温度,微波处理8min,功率为100w,微波处理结束后继续悬浮搅拌24h,抽滤悬浮液,将产物在40℃、常压下干燥至恒重,得到vii晶型甲磺酸达比加群酯。
本实施例所得晶体的hplc纯度为99.15%,表征数据与实施例1相同。
实施例5:
将干燥的甲磺酸达比加群酯晶型i3.27g加入56.42g无水乙醇中形成悬浮液,在搅拌下将该悬浮液恒温于15℃,恒定温度,微波处理10min,功率为200w,微波处理结束后继续悬浮搅拌24h,抽滤悬浮液,将产物在60℃、常压下干燥至恒重,得到vii晶型甲磺酸达比加群酯。
本实施例所得晶体的hplc纯度为99.09%,表征数据与实施例1相同。
实施例6:
将干燥的甲磺酸达比加群酯晶型i2.89g加入57.33g无水乙醇中形成悬浮液,在搅拌下将该悬浮液恒温于15℃,恒定温度,微波处理8min,功率为200w,微波处理结束后继续悬浮搅拌20h,抽滤悬浮液,将产物在60℃、常压下干燥至恒重,得到vii晶型甲磺酸达比加群酯。
本实施例所得晶体的hplc纯度为99.19%,表征数据与实施例1相同。
实施例7:
将干燥的甲磺酸达比加群酯晶型i6.56g加入55.16g无水乙醇中形成悬浮液,在搅拌下将该悬浮液恒温于15℃,恒定温度,微波处理5min,功率为200w,微波处理结束后继续悬浮搅拌10h,抽滤悬浮液,将产物在60℃、常压下干燥至恒重,得到vii晶型甲磺酸达比加群酯。
本实施例所得晶体的hplc纯度为99.22%,表征数据与实施例1相同。
本发明公开和提出的甲磺酸达比加群酯晶型vii及其制备方法,本领域技术人员可通过借鉴本文内容,适当改变原料、工艺参数等环节实现。本发明的方法与产品已通过较佳实施例子进行了描述,相关技术人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和产品进行改动或适当变更与组合,来实现本发明技术。特别需要指出的是,所有相类似的替换和改动对本领域技术人员来说是显而易见的,他们都被视为包括在本发明精神、范围和内容中。
- 还没有人留言评论。精彩留言会获得点赞!