开环异落叶松树脂酚二葡糖苷的制备的制作方法
本申请是2014年6月10日提交的、发明名称为“(s,s)-开环异落叶松树脂酚二葡糖苷和(r,r)-开环异落叶松树脂酚二葡糖苷的制备”的中国专利申请201480041131.2的分案申请。
本发明涉及制备(s,s)-开环异落叶松树脂酚二葡糖苷和(r,r)-开环异落叶松树脂酚二葡糖苷的方法,以及包含其的组合物。
背景技术:
电离辐射对活的生物体产生许多有害作用。人在诊断性和治疗性放射照相程序期间当使用电子设备时会曝露于辐射、曝露于来自核事故背景辅射的辐射,并且在航空航天飞行期间会曝露于辐射。当前全球的发展已经另外建立了恐怖主义,作为可能使许多人可能曝露于致死量辐射的危险手段。因此,极其重要的是鉴别可在曝露于辐射之前施用并且作为放射性曝露之后的治疗施用的放射防护剂。
日益将天然产物和其类似物视作发现和开发放射防护剂的有希望的线索。已经在植物中检测到开环异落叶松树脂酚的(2r,3r)-对映异构体与(2s,3s)]-对映异构体以及内消旋开环异落叶松树脂酚,但是仅(2r,3r)-[(-)开环异落叶松树脂酚]异构体的存在量丰富。虽然这种化合物可从亚麻子中提取得,但是必需的后续纯化是不寻常且冗长的工艺。因此,需要开发一种较短且对映选择性的合成以从容易得到的商业材料制备对映体纯的开环异落叶松树脂酚。
技术实现要素:
本发明提供一种制备式((s,s)-sdg-1)((s,s)-开环异落叶松树脂酚二葡糖苷-1)的化合物的方法
所述方法包括:
(a)使式(6)化合物
与式(7)化合物反应
以制备式((s,s)-s3)化合物
(b)裂解式((s,s)-s3)化合物的苄醚,接着进行分离程序以制备式((s,s)-8)化合物
(c)对式((s,s)-8)化合物脱除保护以制备式((s,s)-sdg-1)化合物。
本发明提供一种制备式((s,s)-sdg-1)化合物的方法
所述方法包括:
(a)使式(5)化合物
与还原剂反应以制备式(s1)化合物
(b)使式(s1)化合物与苄化剂反应以制备式(s2)化合物
(c)使式(s2)化合物与还原剂反应以制备式(6)化合物;
(d)使式(6)化合物与式(7)化合物反应以制备式((s,s)-s3)化合物;
(e)裂解式((s,s)-s3)化合物的苄醚,接着进行分离程序以制备式((s,s)-8)化合物;以及
(f)对式((s,s)-8)化合物脱除保护以制备式((s,s)-sdg-1)化合物。
本发明进一步提供一种制备式((s,s)-sdg-1)化合物的方法,所述方法包括:
(a)使式(s2)化合物与还原剂反应以制备式(6)化合物;
(b)使式(6)化合物与式(7)化合物反应以制备式((s,s)-s3)化合物;
(c)裂解式((s,s)-s3)化合物的苄醚,接着进行分离程序以制备式((s,s)-8)化合物;以及
(d)对式((s,s)-8)化合物脱除保护以制备式((s,s)-sdg-1)化合物。
本发明提供一种制备式((r,r)-sdg-2)((r,r)-开环异落叶松树脂酚二葡糖苷-2)的化合物的方法
所述方法包括:
(a)使式(6)化合物与式(7)化合物反应以制备式((r,r)-s4)化合物;
(b)裂解式((r,r)-s4)化合物的苄醚,接着进行分离程序以制备式((r,r)-9)化合物
(c)对式((r,r)-9)化合物脱除保护以制备式((r,r)-sdg-2)化合物。
本发明进一步提供一种制备式((r,r)-sdg-2)化合物的方法,所述方法包括:
(a)使式(5)化合物与还原剂反应以制备式(s1)化合物;
(b)使式(s1)化合物与苄化剂反应以制备式(s2)化合物;
(c)使式(s2)化合物与还原剂反应以制备式(6)化合物;
(d)使式(6)化合物与式(7)化合物反应以制备式((r,r)-s4)化合物;
(e)裂解式((r,r)-s3)化合物的苄醚,接着进行分离程序以制备式((r,r)-9)化合物;以及
(f)对式((r,r)-9)化合物脱除保护以制备式((r,r)-sdg-2)化合物。
本发明进一步提供一种制备式((r,r)-sdg-2)化合物的方法,所述方法包括:
(a)使式(s2)化合物与还原剂反应以制备式(6)化合物;
(b)使式(6)化合物与式(7)化合物反应以制备式((r,r)-s4)化合物;
(c)裂解式((r,r)-s4)化合物的苄醚,接着进行分离程序以制备式((r,r)-9)化合物;以及
(d)对式((r,r)-9)化合物脱除保护以制备式((r,r)-sdg-2)化合物。
本发明进一步提供一种制备式(6)化合物的方法,所述方法包括使式(s2)化合物与还原剂反应以制备式(6)化合物。
本发明进一步提供一种制备式((s,s)-s3)化合物的方法,所述方法包括使式(6)化合物与式(7)化合物反应以制备式((s,s)-s3)化合物。
本发明进一步提供一种制备式((s,s)-8)化合物的方法,所述方法包括:
(a)使式(6)化合物与式(7)化合物反应以制备式((s,s)-s3)化合物;
(b)裂解式((s,s)-s3)化合物的苄醚,接着进行分离程序以制备式((s,s)-8)化合物。
本发明提供一种制备式((r,r)-s4)化合物的方法,所述方法包括使式(6)化合物与式(7)化合物反应以制备式((r,r)-s4)化合物。
本发明提供一种制备式((r,r)-9)化合物的方法,所述方法包括:
(a)使式(6)化合物与式(7)化合物反应以制备式((r,r)-s4)化合物;
(b)裂解式((r,r)-s4)化合物的苄醚,接着进行分离程序以制备式((r,r)-9)化合物。
本发明进一步提供一种组合物,如药物组合物,其包含式((s,s)-sdg-1)的化合物。
本发明进一步提供一种组合物,如药物组合物,其包含式((r,r)-sdg-2)的化合物。
本发明的其它特征和优势将根据以下详细描述、实施例和图变得而显而易见。然而应了解,所述详细描述和特定实施例尽管指示了本发明的优选实施方案但是仅以说明方式给出,因为在本发明的精神和范围内的各种变化和修改将根据本详细描述变得对本领域技术人员显而易见。
附图说明
图1描绘了二羟基化合物6的ortep图。
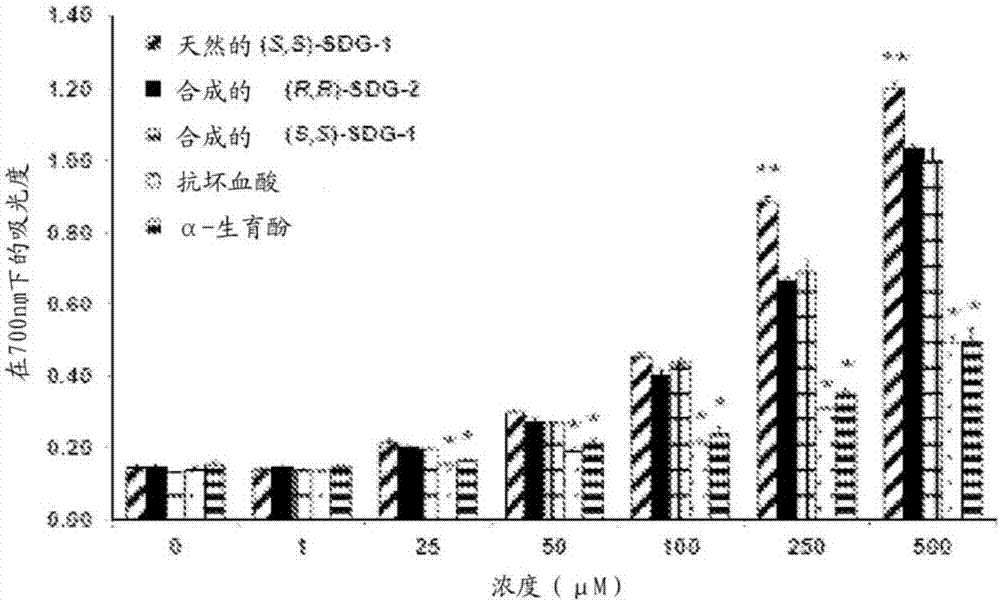
图2说明了合成的(s,s)-sdg-1和(r,r)-sdg-2的还原能力。在700nm下吸光度的增大表明还原能力增大。将结果呈现为平均值±标准偏差(n=3)。*p<0.05,明显低于天然的(s,s)-sdg-1、合成的(r,r)-sdg-2和合成的(s,s)-sdg-1;**p<0.05,明显高于合成的(r,r)-sdg-2、合成的(s,s)-sdg-1、抗坏血酸和α-生育酚。
图3说明了合成的(s,s)-sdg-1和(r,r)-sdg-2的还原能力。反应速率在1-100μm的浓度范围内呈线性。使用线性回归方程以确定ec50。将结果呈现为平均值±标准偏差(n=3)。天然的(s,s)-sdg-1、合成的(r,r)-sdg-2和合成的(s,s)-sdg-1没有彼此显著不同。*p<0.05,显著高于天然的(s,s)-sdg-1、合成的(r,r)-sdg-2和合成的(s,s)-sdg-1。
图4描绘了天然的(s,s)-sdg-1、合成的(s,s)-sdg-1和(r,r)-sdg-2、抗坏血酸和α-生育酚的dpph自由基清除活性。在517nm下dpph吸光度的减小即测量为自由基清除活性。将结果呈现为平均值±标准偏差(n=3)。*p<0.05,显著低于所有其它化合物,**p<0.05,显著低于天然的(s,s)-sdg-1。
图5描绘了天然的(s,s)-sdg-1、合成的(s,s)-sdg-1和(r,r)-sdg-2、抗坏血酸和α-生育酚的dpph自由基清除活性。使用线性回归方程以确定ec50。反应速率在1-100μm的浓度范围内呈线性。将结果呈现为平均值±标准偏差(n=3)。天然的(s,s)-sdg-1、合成的(r,r)-sdg-2、合成的(s,s)-sdg-1,以及α-生育酚没有显著不同。*p<0.05,显著高于天然的(s,s)-sdg-1、合成的(r,r)-sdg-2、合成的(s,s)-sdg-1以及α-生育酚。
图6示出了化合物4的1hnmr(600mhz,cdcl3)和13cnmr(150mhz,cdcl3)谱。
图7示出了化合物5的1hnmr(600mhz,cdcl3)和13cnmr(150mhz,cdcl3)谱。
图8示出了化合物s1的1hnmr(600mhz,cdcl3)和13cnmr(150mhz,cdcl3)谱。
图9示出了化合物s2的1hnmr(600mhz,cdcl3)和13cnmr(150mhz,cdcl3)谱。
图10示出了化合物6的1hnmr(600mhz,cdcl3)和13cnmr(150mhz,cdcl3)谱。
图11示出了化合物1:1(s,s)-s3/(r,r)-s4的1hnmr(600mhz,cdcl3)和13cnmr(150mhz,cdcl3)谱。
图12示出了化合物(s,s)-8的1hnmr(600mhz,cdcl3)和13cnmr(150mhz,cdcl3)谱。
图13示出了化合物(r,r)-9的1hnmr(600mhz,cdcl3)和13cnmr(150mhz,cdcl3)谱。
图14示出了化合物(s,s)-sdg-1的1hnmr(600mhz,cd3od)和13cnmr(150mhz,cd3od)谱。
图15示出了化合物(r,r)-sdg-2的1hnmr(600mhz,cd3od)和13cnmr(150mhz,cd3od)谱。
具体实施方式
本发明提供一种制备式((s,s)-sdg-1)化合物的方法
所述方法包括:
(a)使式(6)化合物
与式(7)化合物反应
以制备式((s,s)-s3)化合物
(b)裂解式((s,s)-s3)化合物的苄醚,接着进行分离程序以制备式((s,s)-8)化合物
(c)对式((s,s)-8)化合物脱除保护以制备式((s,s)-sdg-1)化合物。
在一些实施方案中,所述反应在路易斯酸存在下进行。在某些实施方案中,所述路易斯酸为tmsotf。在一些实施方案中,在活性分子筛存在下进行所述反应。
在一些实施方案中,在meoh中在h2和pd/c存在下进行所述裂解。
在一些实施方案中,使用制备型薄层色谱来进行所述分离程序。
在一些实施方案中,在naome和meoh溶液中进行所述脱除保护。
在一些实施方案中,通过包括以下的方法制备所述式(6)化合物:使式(s2)化合物
与还原剂反应以制备式(6)化合物。在一些实施方案中,所述还原剂为含氢化锂铝(lah)的thf。
在一些实施方案中,通过包括以下的方法制备所述式(s2)化合物:使式(s1)化合物
与苄化剂反应以制备式(s2)化合物。在一些实施方案中,所述苄化剂为bnbr和nah。
在一些实施方案中,通过包括以下的方法制备所述式(s1)化合物:使式(5)化合物
与还原剂反应以制备式(s1)化合物。在一些实施方案中,所述还原剂为h2和pd/c。
在一些实施方案中,通过包括以下的方法制备所述式(5)化合物:使式(4)化合物
与香兰素通过斯陶柏缩合反应来进行反应,接着进行酯化反应以制备式(5)化合物。在一些实施方案中,所述斯陶柏缩合反应在meoh中并且在锂(例如锂线)存在下进行。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
在一些实施方案中,通过包括使香兰素与丁二酸甲酯通过斯陶柏缩合反应进行反应,接着进行酯化反应的方法来制备所述式(4)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
本发明提供一种制备式((s,s)-sdg-1)化合物的方法
所述方法包括:
(a)使式(s2)化合物
与还原剂反应以制备式(6)化合物
(b)使式(6)化合物与式(7)化合物反应
以制备式((s,s)-s3)化合物
(c)裂解式((s,s)-s3)化合物的苄醚,接着进行分离程序以制备式((s,s)-8)化合物
(d)对式((s,s)-8)化合物脱除保护以制备式((s,s)-sdg-1)化合物。
在一些实施方案中,在tmsotf存在下进行步骤(a)的所述反应。在一些实施方案中,在活性分子筛存在下进行步骤(a)的所述反应。
在一些实施方案中,在h2和pd/c存在下在meoh中进行所述裂解。
在一些实施方案中,使用制备型薄层色谱来进行所述分离程序。
在一些实施方案中,在naome和meoh溶液中进行所述脱除保护。
在一些实施方案中,所述还原剂为含氢化锂铝(lah)的thf。
在一些实施方案中,通过包括以下的方法制备所述式(s2)化合物:使式(s1)化合物
与苄化剂反应以生成式(s2)化合物。在一些实施方案中,所述苄化剂为bnbr和nah。
在一些实施方案中,通过包括以下的方法制备所述式(s1)化合物:使式(5)化合物
与还原剂反应以制备式(s1)化合物。在一些实施方案中,所述还原剂为h2和pd/c。
在一些实施方案中,通过包括以下的方法制备所述式(5)化合物:使式(4)化合物
与香兰素通过斯陶柏缩合反应来进行反应,接着进行酯化反应以制备式(5)化合物。
在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。
在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
在一些实施方案中,通过包括使香兰素与丁二酸甲酯通过斯陶柏缩合反应进行反应,接着进行酯化反应的方法来制备所述式(4)化合物。
在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
本发明提供一种制备式((s,s)-sdg-1)化合物的方法
所述方法包括:
(a)使式(5)化合物
与还原剂反应以制备式(s1)化合物
(b)使式(s1)化合物
与苄化剂反应以制备式(s2)化合物
(c)使式(s2)化合物与还原剂反应以制备式(6)化合物
(d)使式(6)化合物与式(7)化合物反应
以制备式((s,s)-s3)化合物
(e)裂解式((s,s)-s3)化合物的苄醚,接着进行分离程序以制备式((s,s)-8)化合物
(f)对式((s,s)-8)化合物脱除保护以制备式((s,s)-sdg-1)化合物。
在一些实施方案中,通过包括以下的方法制备所述式(5)化合物:使式(4)化合物
与香兰素通过斯陶柏缩合反应来进行反应,接着进行酯化反应以制备式(5)化合物。
本发明提供一种制备式((rr)-sdg-2)化合物的方法
所述方法包括:
(a)使式(6)化合物
与式(7)化合物反应
以制备式((r,r)-s4)化合物
(b)裂解式((r,r)-s4)化合物的苄醚,接着进行分离程序以制备式((r,r)-9)化合物
(c)对式((s,s)-9)化合物脱除保护以制备式((r,r)-sdg-2)化合物。
在一些实施方案中,所述反应在路易斯酸存在下进行。在某些实施方案中,所述路易斯酸为tmsotf。
在一些实施方案中,在活性分子筛存在下进行所述反应。
在一些实施方案中,在h2和pd/c存在下在meoh中进行所述裂解。在一些实施方案中,使用制备型薄层色谱来进行所述分离程序。
在一些实施方案中,在naome和meoh溶液中进行所述脱除保护。
在一些实施方案中,通过包括以下的方法制备所述式(6)化合物:使式(s2)化合物
与还原剂反应以制备式(6)化合物。
在一些实施方案中,所述还原剂为含氢化锂铝(lah)的thf。
在一些实施方案中,通过包括以下的方法制备所述式(s2)化合物:使式(s1)化合物
与苄化剂反应以制备式(s2)化合物。在一些实施方案中,所述苄化剂为bnbr和nah。
在一些实施方案中,通过包括以下的方法制备所述式(s1)化合物:使式(5)化合物
与还原剂反应以制备式(s1)化合物。在一些实施方案中,所述还原剂为h2和pd/c。
在一些实施方案中,通过包括以下的方法制备所述式(5)化合物:使式(4)化合物
与香兰素通过斯陶柏缩合反应来进行反应,接着进行酯化反应以制备式(5)化合物。
在一些实施方案中,所述斯陶柏缩合反应在meoh中并且在锂(例如锂线)存在下进行。
在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
在一些实施方案中,通过包括使香兰素与丁二酸甲酯通过斯陶柏缩合反应进行反应,接着进行酯化反应的方法来制备所述式(4)化合物。
在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。
在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
本发明提供一种制备式((r,r)-sdg-2)化合物的方法
所述方法包括:
(a)使式(s2)化合物
与还原剂反应以制备式(6)化合物
(b)使式(6)化合物与式(7)化合物反应
以制备式((r,r)-s4)化合物
(c)裂解式((r,r)-s4)化合物的苄醚,接着进行分离程序以制备式((r,r)-9)化合物
(d)对式((r,r)-9)化合物脱除保护以制备式((r,r)-sdg-2)化合物。
在一些实施方案中,在tmsotf存在下进行步骤(a)的所述反应。在一些实施方案中,在活性分子筛存在下进行步骤(a)的所述反应。
在一些实施方案中,在h2和pd/c存在下在meoh中进行所述裂解。在一些实施方案中,使用制备型薄层色谱来进行所述分离程序。
在一些实施方案中,在naome和meoh溶液中进行所述脱除保护。
在一些实施方案中,所述还原剂为含氢化锂铝(lah)的thf。
在一些实施方案中,通过包括以下的方法制备所述式(s2)化合物:使式(s1)化合物
与苄化剂反应以生成式(s2)化合物。
在一些实施方案中,所述苄化剂为bnbr和nah。
在一些实施方案中,通过包括以下的方法制备所述式(s1)化合物:使式(5)化合物
与还原剂反应以制备式(s1)化合物。
在一些实施方案中,所述还原剂为h2和pd/c。
在一些实施方案中,通过包括以下的方法制备所述式(5)化合物:使式(4)化合物
与香兰素通过斯陶柏缩合反应来进行反应,接着进行酯化反应以制备式(5)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
在一些实施方案中,通过包括使香兰素与丁二酸甲酯通过斯陶柏缩合反应进行反应,接着进行酯化反应的方法来制备所述式(4)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
本发明提供一种制备式((r,r)-sdg-2)化合物的方法
所述方法包括:
(a)使式(5)化合物
与还原剂反应以制备式(s1)化合物
(b)使式(s1)化合物
与苄化剂反应以制备式(s2)化合物
(c)使式(s2)化合物与还原剂反应以制备式(6)化合物
(d)使式(6)化合物与式(7)化合物反应
以制备式((r,r)-s4)化合物
(e)裂解式((r,r)-s3)化合物的苄醚,接着进行分离程序以制备式((r,r)-9)化合物
(f)对式((r,r)-9)化合物脱除保护以制备式((r,r)-sdg-2)化合物。
在一些实施方案中,通过包括以下的方法制备所述式(5)化合物:使式(4)化合物
与香兰素通过斯陶柏缩合反应来进行反应,接着进行酯化反应以制备式(5)化合物。
本发明提供一种制备式(6)化合物的方法
所述方法包括使式(s2)化合物
与还原剂反应以制备式(6)化合物。在一些实施方案中,所述还原剂为含氢化锂铝(lah)的thf。
在一些实施方案中,通过包括以下的方法制备所述式(s2)化合物:使式(s1)化合物
与苄化剂反应以制备式(s2)化合物。在某些实施方案中,所述苄化剂为bnbr和nah。
在一些实施方案中,通过包括以下的方法制备所述式(s1)化合物:使式(5)化合物
与还原剂反应以制备式(s1)化合物。在某些实施方案中,所述还原剂为h2和pd/c。
在一些实施方案中,通过包括以下的方法制备所述式(5)化合物:使式(4)化合物
与香兰素通过斯陶柏缩合反应来进行反应,接着进行酯化反应以制备式(5)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
在一些实施方案中,通过包括使香兰素与丁二酸甲酯通过斯陶柏缩合反应进行反应,接着进行酯化反应的方法来制备所述式(4)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
本发明提供一种制备式((s,s)-s3)化合物的方法
所述方法包括使式(6)化合物
与式(7)化合物反应
以制备式((s,s)-s3)化合物。
在一些实施方案中,所述反应在路易斯酸存在下进行。在一些实施方案中,所述路易斯酸为tmsotf。在一些实施方案中,在活性分子筛存在下进行所述反应。
在一些实施方案中,通过包括以下的方法制备所述式(6)化合物:使式(s2)化合物
与还原剂反应以制备式(6)化合物。在一些实施方案中,所述还原剂为含氢化锂铝(lah)的thf。
在一些实施方案中,通过包括以下的方法制备所述式(s2)化合物:使式(s1)化合物
与苄化剂反应以制备式(s2)化合物。在某些实施方案中,所述苄化剂为bnbr和nah。
在一些实施方案中,通过包括以下的方法制备所述式(s1)化合物:使式(5)化合物
与还原剂反应以制备式(s1)化合物。在某些实施方案中,所述还原剂为h2和pd/c。
在一些实施方案中,通过包括以下的方法制备所述式(5)化合物:使式(4)化合物
与香兰素通过斯陶柏缩合反应来进行反应,接着进行酯化反应以制备式(5)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
在一些实施方案中,通过包括使香兰素与丁二酸甲酯通过斯陶柏缩合反应进行反应,接着进行酯化反应的方法来制备所述式(4)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
本发明提供一种制备式((s,s)-8)化合物的方法
所述方法包括:
(a)使式(6)化合物
与式(7)化合物反应
以制备式((s,s)-s3)化合物
(b)裂解式((s,s)-s3)化合物的苄醚,接着进行分离程序以制备式((s,s)-8)化合物。
在一些实施方案中,在tmsotf存在下进行所述反应。在一些实施方案中,反应在活性分子筛存在下进行。在一些实施方案中,在h2和pd/c存在下在meoh中进行所述裂解。在一些实施方案中,使用制备型薄层色谱来进行所述分离程序。
在一些实施方案中,通过包括以下的方法制备所述式(6)化合物:使式(s2)化合物
与还原剂反应以制备式(6)化合物。在某些实施方案中,所述还原剂为含氢化锂铝(lah)的thf。
在一些实施方案中,通过包括以下的方法制备所述式(s2)化合物:使式(s1)化合物
与苄化剂反应以制备式(s2)化合物。在某些实施方案中,所述苄化剂为bnbr和nah。
在一些实施方案中,通过包括以下的方法制备所述式(s1)化合物:使式(5)化合物
与还原剂反应以制备式(s1)化合物。在某些实施方案中,所述还原剂为h2和pd/c。
在一些实施方案中,通过包括以下的方法制备所述式(5)化合物:使式(4)化合物
与香兰素通过斯陶柏缩合反应来进行反应,接着进行酯化反应以制备式(5)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
在一些实施方案中,通过包括使香兰素与丁二酸甲酯通过斯陶柏缩合反应进行反应,接着进行酯化反应的方法来制备所述式(4)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
本发明提供一种制备式((r,r)-s4)化合物的方法
所述方法包括:
(a)使式(6)化合物
与式(7)化合物反应
以制备式((r,r)-s4)化合物。
在一些实施方案中,在tmsotf存在下进行所述反应。
在一些实施方案中,在活性分子筛存在下进行所述反应。
在一些实施方案中,通过包括以下的方法制备所述式(6)化合物:使式(s2)化合物
与还原剂反应以制备式(6)化合物。在某些实施方案中,所述还原剂为含氢化锂铝(lah)的thf。
在一些实施方案中,通过包括以下的方法制备所述式(s2)化合物:使式(s1)化合物
与苄化剂反应以制备式(s2)化合物。在一些实施方案中,所述苄化剂为bnbr和nah。
在一些实施方案中,通过包括以下的方法制备所述式(s1)化合物:使式(5)化合物
与还原剂反应以制备式(s1)化合物。在某些实施方案中,所述还原剂为h2和pd/c。
在一些实施方案中,通过包括以下的方法制备所述式(5)化合物:使式(4)化合物
与香兰素通过斯陶柏缩合反应来进行反应,接着进行酯化反应以制备式(5)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
在一些实施方案中,通过包括使香兰素与丁二酸甲酯通过斯陶柏缩合反应进行反应,接着进行酯化反应的方法来制备所述式(4)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
本发明提供一种制备式((r,r)-9)化合物的方法
所述方法包括:
(a)使式(6)化合物
与式(7)化合物反应
以制备式((r,r)-s4)化合物
(b)裂解式((r,r)-s4)化合物的苄醚,接着进行分离程序以制备式((r,r)-9)化合物。
在一些实施方案中,在tmsotf存在下进行所述反应。在一些实施方案中,在活性分子筛存在下进行所述反应。在一些实施方案中,在h2和pd/c存在下在meoh中进行所述裂解。在一些实施方案中,使用制备型薄层色谱来进行所述分离程序。
在一些实施方案中,通过包括以下的方法制备所述式(6)化合物:使式(s2)化合物
与还原剂反应以制备式(6)化合物。在某些实施方案中,所述还原剂为含氢化锂铝(lah)的thf。
在一些实施方案中,通过包括以下的方法制备所述式(s2)化合物:使式(s1)化合物
与苄化剂反应以制备式(s2)化合物。在某些实施方案中,所述苄化剂为bnbr和nah。
在一些实施方案中,通过包括以下的方法制备所述式(s1)化合物:使式(5)化合物
与还原剂反应以制备式(s1)化合物。在某些实施方案中,所述还原剂为h2和pd/c。
在一些实施方案中,通过包括以下的方法制备所述式(5)化合物:使式(4)化合物
与香兰素通过斯陶柏缩合反应来进行反应,接着进行酯化反应以制备式(5)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
在一些实施方案中,通过包括使香兰素与丁二酸甲酯通过斯陶柏缩合反应进行反应,接着进行酯化反应的方法来制备所述式(4)化合物。在一些实施方案中,在meoh中并且在锂线存在下进行所述斯陶柏缩合反应。在一些实施方案中,用meoh在h2so4存在下进行所述酯化反应。
术语“反应”意指使指定试剂以允许其根据化学体系的热力学和动力学而发生分子间相互作用和化学转变的方式在一起。特别对于固体试剂来说,可通过使用适当的溶剂或者至少一种试剂至少部分可溶的溶剂混合物来帮助反应。反应典型地进行合适次数并且在适于产生所需化学转变的条件下进行。
本文所述的方法可以包括其它合适的经历上述合成途径的起始物质。在一些实施方案中,本文所述的方法还可以在上述步骤之前或之后包括额外步骤,以添加或去除合适的保护基。另外,各种合成步骤可以用交替的顺序或次序进行以得到所需化合物。
一方面,本发明提供一种以可商购获得的化合物例如香兰素作为起始物来制备(s,s)-开环异落叶松树脂酚二葡糖苷((s,s)-sdg-1)与(r,r)-开环异落叶松树脂酚二葡糖苷((r,r)-sdg-2))的简明途径。化合物((s,s)-sdg-1)和((r,r)-sdg-2)为亚麻子中的生物活性成分。这两种化合物对羟基、过氧化氢和dpph(2,2-二苯基-1-苦味基偕腙肼)自由基都具有强的还原能力和高的自由基清除活性。
本发明的方法可得到大致上纯的化合物(s,s)-开环异落叶松树脂酚二葡糖苷((s,s)-sdg-1)和化合物(r,r)-开环异落叶松树脂酚二葡糖苷((r,r)-sdg-2)。对于((s,s)-sdg-1),“大致上纯”意指化合物((s,s)-sdg-1)至少大致上与其形成或检测到的环境分离。大致的纯度可包括含有至少约80.0重量%,或至少约85.0重量%,或至少约90.0重量%,或至少约95.0重量%,或至少约97.0重量%,或至少约98.0重量%,或至少约99.0重量%,或至少约99.2重量%,或至少约99.4重量%,或至少约99.6重量%,或至少约99.8重量%,或至少约99.9重量%,或甚至约100重量%所述化合物的组合物。对于((r,r)-sdg-2),“大致上纯”意指化合物((r,r)-sdg-2)至少大致上与其形成或检测到的环境分离。大致的纯度可包括含有至少约80.0重量%,或至少约85.0重量%,或至少约90.0重量%,或至少约95.0重量%,或至少约97.0重量%,或至少约98.0重量%,或至少约99.0重量%,或至少约99.2重量%,或至少约99.4重量%,或至少约99.6重量%,或至少约99.8重量%,或至少约99.9重量%,或甚至约100重量%所述化合物的组合物。
“药物组合物”是呈适用于向受试者施用的形式的含有化合物的制剂。在一些实施方案中,药物组合物是散装的或呈单位剂型。单位剂型是多种形式包括例如胶囊、iv袋、片剂、气雾剂吸入器上的单泵或小瓶中的任一种。单位剂量组合物中活性成分(例如本发明的化合物或其盐、溶剂合物、多形体或前药的制剂)的量是有效量并且根据所涉及的具体治疗而变。本领域技术人员应了解,有时有必要根据患者年龄和状况对剂量进行常规改变。剂量还将取决于施用途径。涵盖多种途径,包括口服、经肺、经直肠、胃肠外、透皮、皮下、静脉内、肌肉内、腹膜内、吸入、经颊、舌下、胸膜内、鞘内、鼻内等。本发明的化合物的表面或透皮施用的剂型包括散剂、喷雾剂、软膏剂、膏剂、霜剂、洗剂、凝胶剂、溶液、贴剂和吸入剂。在一些实施方案中,使活性化合物在无菌条件下与药学上可接受的载体混合,并且与任何需要的防腐剂、缓冲剂或推进剂混合。
“受试者”包括哺乳动物,例如人、伴侣动物(例如狗、猫、鸟等)、农畜(例如奶牛、绵羊、猪、马、家禽等)和实验动物(例如大鼠、小鼠、豚鼠、鸟等)。在一些实施方案中,受试者是人。
如本文所用,词语“药学上可接受的”是指在合理的医学判断的范围内、适用于与人畜组织接触而无过量毒性、刺激、过敏反应或其它问题或并发症、与合理的益处/风险比相称的那些化合物、物质、组合物、载体和/或剂型。
“药学上可接受的赋形剂”意指适用于制备一般为安全无毒并且在生物学上和其它方面都没有不合需要的药物组合物的赋形剂,并且包括可为兽医之用以及人药物之用所接受的赋形剂。如本文所用的“药学上可接受的赋形剂”包括一种与一种以上这类赋形剂。
本发明的实施方案还包括包含(s,s)-开环异落叶松树脂酚二葡糖苷((s,s)-sdg-1)的组合物。优选地,这些组合物是包含(s,s)-开环异落叶松树脂酚二葡糖苷((s,s)-sdg-1)和至少一种药学上可接受的赋形剂的药物组合物。在一些实施方案中,可以用大致上纯的(s,s)-开环异落叶松树脂酚二葡糖苷((s,s)-sdg-1)制备所述组合物和药物组合物。在一些实施方案中,所述组合物和药物组合物的非对映异构体过量(de)为至少90%de、优选地至少95%de、更优选地至少98%de并且甚至更优选地至少99%de,并且最优选地约100%de。组合物和药物组合物还可以制备成化合物的非对映异构体形式的混合物(例如作为外消旋混合物或作为((s,s)-sdg-1)相对于((r,r)-sdg-2)比率为60:40、70:30、80:20或90:10的混合物)。
本发明的实施方案还包括包含(r,r)-开环异落叶松树脂酚二葡糖苷((r,r)-sdg-2)的组合物。优选地,这些组合物是包含(r,r)-开环异落叶松树脂酚二葡糖苷((r,r)-sdg-2)和至少一种药学上可接受的赋形剂的药物组合物。在一些实施方案中,可以用大致上纯的(r,r)-开环异落叶松树脂酚二葡糖苷((r,r)-sdg-2)制备所述组合物和药物组合物。在一些实施方案中,所述组合物和药物组合物的非对映异构体过量(de)为至少90%de、优选地至少95%de、更优选地至少98%de并且甚至更优选地至少99%de,并且最优选地约100%de。组合物和药物组合物还可以制备成化合物的非对映异构体形式的混合物(例如作为外消旋混合物或作为((r,r)-sdg-2)相对于((s,s)-sdg-1)比率为60:40、70:30、80:20或90:10的混合物)。
在整个说明书中,当将组合物描述为具有、包括或包含特定组分时,涵盖所述组合物还基本上由所述组分组成或者由所述组分组成。类似地,当将方法描述为具有、包括或包含特定方法步骤时,所述方法还基本上由所述处理步骤组成或者由所述处理步骤组成。进一步,应了解,步骤的顺序或实施某些动作的顺序并不重要,只要本发明保持可操作即可。此外,可以同时进行两个或更多个步骤或动作。
呈现以下实施例以更完整地说明本发明的优选实施方案。然而,决不应将其视为限制本发明的广泛范围。
实施例
化学反应的通用程序
除非另作说明,否则所有反应都在氩气气氛下、在无水溶剂下、在无水条件下进行。通过使可商购获得的预先干燥的不含氧制剂穿过活性氧化铝柱来获得无水四氢呋喃(thf)、二甲基甲酰胺(dmf)和二氯甲烷(ch2cl2)。除非另有说明,否则所得物(yields)是指在色谱和分光(1hnmr)上均匀的物质。除非另有说明,否则试剂是以最高的商品质量购得并且在没有进一步纯化下便加以使用。通过在0.25mme.merck硅胶板(60f-254)上进行的薄层色谱(tlc)、使用紫外光作为可视化剂并且使用磷钼酸和硫酸铈的乙醇溶液,以及热作为显影剂来监测反应。将e.merck硅胶(60,粒度0.040-0.063mm)用于快速柱色谱。除非另作说明,否则在0.25或0.50mme.merck硅胶板(60f-254)上进行制备型薄层色谱(ptlc)分离。在brukerdrx-600仪器上记录nmr谱并且使用残余未氘化的溶剂作为内部参考来进行校准。使用以下缩写来解释多峰性:s=单峰、d=双重峰、t=三重峰、m=多重峰。在perkin-elmerspectrum100ft-ir光谱计上记录ir谱。在使用esi(电喷雾电离)的vgzab-zse质谱仪上记录高分辨率质谱(hr-ms)。
方案1示出了用于合成化合物(6),即用于制备(s,s)-开环异落叶松树脂酚二葡糖苷和(r,r)-开环异落叶松树脂酚二葡糖苷的中间化合物的反应途径。
方案1:合成化合物(6)
实施例1:合成酚4
在控制放热量的冰浴下,向香兰素3(14.0g,92.1mmol,1当量)和丁二酸二甲酯(12.13ml,92.1mmol,1当量)于meoh(450ml)中的溶液中逐片缓慢添加锂线(1.79g,257.9mmol,2.8当量)。在初始的锂已经完全溶解之后,再缓慢逐片添加锂(1.98g,285.5mmol,3.1当量)并且搅拌直到完全溶解为止。然后在回流下加热反应混合物48小时。在将溶液冷却到室温之后,通过旋蒸浓缩来去除大部分甲醇。添加etoac(1000ml)并且溶液用2m盐酸水溶液(700ml)、h2o(3×1000ml)、然后用盐水(200ml)洗涤。有机层然后被干燥(mgso4)、过滤和浓缩。将粗物质溶解于meoh(230ml)中,添加h2so4(1ml),并且在回流下加热溶液隔夜。在次日早晨冷却时,添加nahco3(3.0g)以淬灭h2so4并且溶液被大部分旋蒸浓缩。添加etoac(500ml),并且溶液用h2o(2×200ml)、然后用盐水(100ml)洗涤。有机层然后被干燥(mgso4)、过滤并且浓缩为棕色油状物。溶解于少量ch2cl2中,然后进行快速柱色谱(硅石,1:9→2:8→3:7→4:6→5:5乙醚:己烷),提供呈灰白色固体状的4(18.0g,64.2mmol,70%收率,烯烃几何形状未指定)。4:rf=0.17(硅石,乙醚:己烷1:1);ir(膜):νmax=3422,1702,1514,1434,1258,11951159,1093,1030,924,821770,729cm-1;1hnmr(600mhz,cdcl3)δ=7.79(s,1h),6.90-6.85(m,3h),6.19(s,1h),3.82(s,3h),3.78(s,3h),3.69(s,3h),3.57(s,2h)ppm;13cnmr(150mhz,cdcl3)δ=171.94,168.13,146.80,146.61,142.38,126.96,123.39,123.31,114.76,111.80,55.85,52.24,52.20,33.61ppm;hrms(esi-tof):c14h16o6[m+h+]的计算值280.102,实验值281.1022。
实施例2:合成二酯5
在控制放热量的冰浴下,向二酯4(15.00g,53.52mmol,1当量)和香兰素3(8.14g,53.52mmol,1当量)于meoh(200ml)中的溶液中逐片缓慢添加锂线(2.6g,374.6mmol,7当量),并且搅拌直到完全溶解为止。然后在回流下加热反应混合物48小时。在将溶液冷却到室温之后,通过旋蒸浓缩来去除大部分甲醇。添加etoac(200ml),溶液用2m盐酸水溶液(500ml)酸化,然后用etoac(2×200ml)萃取。合并的有机物用h2o(2×200ml)、然后用盐水(200ml)洗涤、并且干燥(mgso4)、过滤并浓缩。将残留物溶解于meoh(300ml)中、添加浓h2so4(1ml),并且在回流下加热溶液隔夜。在次日早晨冷却时,添加nahco3(3.0g)以淬灭h2so4并且溶液被大部分旋蒸浓缩。添加etoac(400ml),并且溶液用h2o(2×200ml)、然后用盐水(100ml)洗涤。有机层然后被干燥(mgso4)、过滤和浓缩。溶解于少量ch2cl2中,然后进行快速柱色谱(硅石,2:1:7→3:1:6→4:1:5etoac:ch2cl2:己烷)提供呈黄橙色固体状的5(13.6g,32.8mmol,61%收率,烯烃几何形状未指定)。5:rf=0.28(硅石,etoac:己烷1:1);ir(膜):νmax=3388,1696,1588,1509,1431,1209,1157,1029,817,733cm-1;1hnmr(600mhz,cdcl3)δ=7.86(s,2h),7.11(d,j=1.64hz,2h),7.04(dd,j=8.48,1.72hz,2h),6.82(d,j=8.38hz,2h),6.08(s,2h),3.7(s,6h),3.69(s,6h)ppm;13cnmr(150mhz,cdcl3)δ=168.00,147.59,146.54,142.64,127.10,125.38,124.06,114.71,111.52,55.77,52.51ppm;hrms(esi-tof):c22h22o8[m+h+]的计算值415.1387,实验值415.1378。
实施例3:合成二酯s1
通过短暂暴露于真空并且用氩气回填几次使5(5.00g,12.07mmol,1当量)于meoh(120ml)中的溶液饱和含有氩气气氛。添加钯/碳(10重量%钯,0.500g)并且通过真空/回填h2使溶液饱和含有h2气氛。搅拌溶液隔夜。次日早晨将溶液放在氩气气氛下,添加ch2cl2(400ml),并且允许溶液搅拌1小时。然后使混合物通过硅藻土垫(1.5in,用meoh和ch2cl2洗涤)过滤,并且浓缩滤液。将所得固体溶解于少量ch2cl2中并且通过快速柱色谱(2:8:1→3:7:1→4:6:1etoac:己烷:ch2cl2)纯化以得到呈灰白色固体状的s1(4.24g,10.1mmol,84%)。s1:rf=0.24(硅石,etoac:己烷:ch2cl24:6:1);ir(膜):νmax=3440,2951,1726,1514,1432,1267,1198,1151,1121,1029,817,797cm-1;1hnmr(600mhz,cdcl3)δ=6.78(d,j=8.09hz,2h),6.58(d,j=8.09hz,2h),6.45(s,2h),5.65(s,2h),3.75(s,6h),3.64(s,6h),3.00-2.94(m,2h),2.92-2.82(m,4h)ppm;13cnmr(150mhz,cdcl3)δ=174.03,146.49,144.21,130.41,121.88,114.18,111.26,55.73,51.86,47.67,35.38ppm;hrms(esi-tof):c22h26o8[m+h+]的计算值419.17,实验值419.1700。
实施例4:合成苄醚s2
向用冰浴冷却到0℃的s1(1.00g,2.39mmol,1当量)于dmf(24ml)中的溶液中缓慢添加nah(0.201g,5.02mmol,60%分散液,2.1当量)并且在0℃下搅拌溶液1小时。历经1分钟添加bnbr(910μl,7.65mmol,3.2当量),并且在0℃下搅拌溶液4小时。然后将反应混合物倾入h2o(300ml)和etoac(100ml)中。用h2o(3×200ml)和盐水(1×50ml)洗涤有机层、干燥(mgso4)、过滤和浓缩。将所得固体溶解于少量ch2cl2中并且通过快速柱色谱(硅石,9:1:1→8:2:1己烷:etoac:ch2cl2)纯化以得到呈白色固体状的s2(1.36g,2.27,95%收率)。s2:rf=0.23(硅石,etoac:己烷3:7);ir(膜):νmax=2949,1730,1512,1453,1253,1225,1138,1023,733cm-1;1hnmr(600mhz,cdcl3)δ=7.45-7.40(m,4h),7.38-7.33(m,4h),7.31-7.27(m,2h),6.74(d,j=8.12hz,2h),6.59(d,j=1.69hz,2h),6.53(dd,j=8.12,1.69hz,2h),5.12(s,4h),3.80(s,6h),3.61(s,6h),3.03-2.85(m,6h)ppm;13cnmr(150mhz,cdcl3)δ=174.00,149.58,146.87,137.37,131.74,128.65,127.93,127.37,121.09,114.05,112.73,71.14,56.00,51.91,47.91,35.32ppm;hrms(esi-tof):c36h38o8[m+h+]的计算值599.2639,实验值599.2651。
实施例5:合成二醇6
在0℃下向s2(0.341g,0.570mmol,1当量)于thf(6ml)中的溶液中逐滴添加氢化锂铝(thf中的1m溶液,1.14ml,1.14mmol,2当量)。允许溶液达到25℃隔夜。将所得混合物倾入含有h2o(200ml)和etoac(100ml)的烧瓶中。然后添加50ml饱和罗谢尔盐水溶液并且搅拌混合物直到各层可容易地分离为止。然后用h2o(2×100ml)和盐水(1×50ml)洗涤有机层,并且干燥(mgso4)、过滤和浓缩。将所得固体溶解于少量ch2cl2中并且通过快速柱色谱(硅石,5:5:1→7:3:1→8:2:1etoac:己烷:ch2cl2)纯化以得到呈白色固体状的二醇6(0.286g,0.527mmol,93%收率)。6:rf=0.2(硅石,6:4etoac:己烷);ir(膜):νmax=3287,2933,1510,1453,1259,1223,1137,1009,733,695cm-1;1hnmr(600mhz,cdcl3)δ=7.47-7.42(m,4h),7.40-7.34(m,4h),7.33-7.28(m,2h),6.78(d,j=8.15hz,2h),6.69(s,2h),6.62(d,j=8.15hz,2h),5.11(s,4h),4.09(s,2h),3.82(s,6h),3.78(d,j=11.07hz,2h),3.49(d,j=11.07hz,2h),2.80-2.72(m,2h),2.69-2.62(m,2h),1.86(s,2h)ppm;13cnmr(150mhz,cdcl3)δ149.47,146.38,137.29,133.87,128.51,127.80,127.28,121.02,114.02,112.79,71.12,60.19,55.96,43.79,35.81ppm;hrms(esi-tof):c34h38o6[m+h+]的计算值543.2741,实验值543.2741。
方案2示出了用于以化合物(6)作为起始物来合成(s,s)-开环异落叶松树脂酚二葡糖苷和(r,r)-开环异落叶松树脂酚二葡糖苷的反应途径。
方案2:从化合物(6)合成((s,s)-sdg-1)和((r,r)-sdg-2)
实施例6:合成苷化异构体(s,s)-s3和(r,r)-s4
向烧瓶中装入二醇6(0.408g,0.751mmol,1当量)和三氯乙酰亚胺酸酯73(1.67g,2.25mmol,3当量),并且通过苯共沸物(3×10ml)干燥。添加活性
实施例7:酚(s,s)-8和(r,r)-9
通过短暂暴露于真空并且用氩气回填几次使(s,s)-s3与(r,r)-s4(0.612g,0.360mmol,1当量)于etoac(3.6ml)中的1:1混合物饱和含有氩气气氛。添加钯/碳(10重量%钯,0.120g)并且通过真空/回填h2使溶液饱和含有h2。在25℃下搅拌36小时之后,将溶液放在氩气气氛下,通过硅藻土垫(1.5in,用etoac和ch2cl2洗涤)过滤,并且浓缩滤液。将所得固体溶解于少量ch2cl2中并且通过快速柱色谱(硅石,3:7→4:6→5:5etoac:己烷)纯化以得到呈非对映异构体的1:1混合物的脱苄基化糖苷(0.470g,0.309mmol,86%收率)。可通过制备型薄层色谱(硅石,2mm,多块板,7:20etoac:己烷,>10次洗脱运行)分离非对映异构体以得到呈灰白色固体状的(s,s)-8和(r,r)-9。(s,s)-8:rf=0.10(4:6etoac:己烷);[α]d32=+1.2(etoac,c=4.3);ir(膜):νmax=3460,2938,1729,1514,1451,1265,1023,1061,1027,709cm-1;1hnmr(600mhz,cdcl3)δ=8.03(d,j=8.42hz,4h),7.92(d,j=8.22hz,4h),7.83(d,j=7.48hz,8h),7.58-7.46(m,6h),7.45-7.27(m,18h),6.62(d,j=7.90hz,2h),6.43(dd,j=7.90,1.16hz,2h),6.38(d,j=1.16hz,2h),5.80(t,j=9.57hz,2h),5.64(t,j=9.57hz,2h),5.44(t,j=9.74hz,2h),5.40(s,2h),4.62(dd,j=12.09,2.98hz,2h),4.44(dd,j=12.09,5.17hz,2h),4.41(d,j=8.03hz,2h),3.96(m,2h),3.69(s,6h),3.64(dd,j=9.38,2.82hz,2h),3.21(dd,j=9.43,4.13hz,2h),2.48(d,j=6.8hz,4h),1.71(s,2h)ppm;13cnmr(150mhz,cdcl3)δ=166.26,165.92,165.35,165.05,146.27,143.64,133.60,133.49,133.38,133.37,132.41,129.95,129.88,129.85,129.78,129.67,129.30,128.94,128.60,128.57,128.45,122.01,113.83,111.49,101.37,72.96,72.15,72.00,69.89,69.54,63.16,55.82,40.87,35.26ppm;hrms(esi-tof):c88h78o24[m+h+]的计算值1519.4956,实验值1519.4937.(r,r)-9:rf=0.10(4:6etoac:己烷);[α]d32=+4.8(etoac,c=4.1);ir(膜):νmax=3457,2942,1726,1262,1026,707cm-1;1hnmr(600mhz,cdcl3)δ=8.00(d,j=7.62hz,4h),7.97(d,j=7.71hz,4h),7.93(d,j=7.98hz,4h),7.62(d,j=7.71hz,4h),7.56-7.27(m,24h),6.45(d,j=7.95hz,2h),6.29(s,2h),6.24(d,j=7.95hz,2h),5.91(t,j=9.82hz,2h),5.70(t,j=9.67hz,2h),5.54(t,j=9.23hz,2h),5.40(s,2h),4.71(dd,j=11.87,3.29hz,2h),4.62(d,j=7.92hz,2h),4.42(dd,j=12.14,4.72hz,2h),4.11-4.06(m,2h),3.85(dd,j=9.53,3.46hz,2h),3.65(s,6h),3.34(dd,j=9.70,4.68hz,2h),2.47-2.40(m,2h),2.37-2.30(m,2h),1.82(s,2h)ppm;13cnmr(150mhz,cdcl3)δ=166.23,165.95,165.34,165.21,146.33,143.54,133.56,133.50,133.36,133.30,132.44,129.96,129.92,129.87,129.65,129.42,128.98,128.66,128.55,128.44,121.98,113.66,111.12,101.25,73.07,72.15,72.08,69.88,69.78,62.97,55.78,40.67,35.50ppm;hrms(esi-tof):c36h38o8[m+h+]的计算值1519.4956,实验值1519.4947。
实施例8:合成开环异落叶松树脂酚二葡糖苷(s,s)-sdg-1
向含有无水(s,s)-8(0.043g,0.028mmol,1当量)的烧瓶中添加新鲜制备的naome于meoh中的溶液(0.4m,2ml,28当量),并且在25℃下搅拌溶液60小时。然后使溶液通过硅石垫(1.5in,用meoh洗涤)过滤,并且浓缩滤液。通过制备型薄层色谱(硅石,2mm,9:1→7:3ch2cl2:meoh,然后一半板长为5:5ch2cl2:meoh)纯化所得固体,然后使其穿过逆相硅石小塞(
实施例9:合成开环异落叶松树脂酚二葡糖苷(r,r)-sdg-2
向含有无水(r,r)-9(0.041g,0.027mmol,1当量)的烧瓶中添加新鲜制备的naome于meoh中的溶液(0.4m,2ml,28当量),并且在25℃下搅拌溶液60小时。然后使溶液通过硅石垫(1.5in,用meoh洗涤)过滤,并且浓缩滤液。通过制备型薄层色谱(硅石,2mm,9:1→7:3ch2cl2:meoh,然后一半板长为5:5ch2cl2:meoh)纯化所得固体,然后使其穿过逆相硅石小塞(
生物分析方法
分析试剂盒
从oxfordbiomedicalresearch购得horac分析试纸盒(#ta30)。从cellbiolab,sandiego,ca获得orac分析试纸盒(#sta345)。从chromadex公司,sandiego,ca购得sdg标准物。
还原能力活性分析
如yen和der(j.am.oilchem.soc.1993,70,383)所述进行还原能力的测定。还原能力分析测定了测试化合物的还原电位,所述测试化合物会与铁氰化钾(fe+3)反应形成亚铁氰化钾(fe+2),而亚铁氰化钾随后与氯化铁反应产生在700nm下具有最大吸收的三价铁-二价铁络合物。将各种浓度(1-500μm)的测试化合物溶解于96孔微板中的磷酸钠缓冲液(0.1m,ph6.6)中并且与铁氰化钾(1%)混合。在50℃下孵育样本并且添加等体积的10%三氯乙酸。使上层与去离子水(1:1:2)和氯化铁(0.1%)混合。在700nm下在bio-rad微板读取器(bio-rad,hercules,ca)上读取吸光度。吸光度增大表明还原能力增大。
羟基自由基清除潜力(horac分析)
使用获自oxfordbiomedicalresearch的horac分析试纸盒(#ta30)来评价sdg清除化学体系中的羟基自由基的能力。通过fenton反应从过氧化氢产生羟基自由基。在荧光微板读取器上测量荧光素的氧化。抗氧化剂会抑制荧光素氧化。将没食子酸用作校准曲线的标准物。计算使用符合校准曲线的线性部分的sdg浓度。使用在8μm-1mm范围内的sdg浓度。将针对羟基自由基的抗氧化能力表示为没食子酸当量(gae)。
过氧化氢自由基清除潜力(orac分析)
使用获自cellbiolab(sandiego,ca)的orac分析试纸盒(#sta345)来评价sdg清除化学体系中的过氧化氢自由基的能力。由aaph(2,2'-偶氮双(2-甲脒基丙烷)二盐酸盐)产生过氧化氢自由基。使用荧光微板读取器测量荧光素的氧化。抗氧化剂会抑制荧光素氧化。将trolox用作校准曲线的标准物。计算使用符合校准曲线的线性部分的sdg浓度。使用在8μm-1mm范围内的sdg浓度。将针对过氧化氢自由基的抗氧化能力表示为trolox当量(te)。
dpph自由基清除分析。
由moree等(freerad.antiox.2011,1,31)所述,对微板使用较小修改来评估sdg清除dpph自由基的能力。简言之,将不同浓度的sdg异构体和其它测试化合物与200μl培养基在含有0.1mtris缓冲液(ph7.4)和250μmdpph溶液的96孔微板中一起孵育,并且在黑暗中保持20分钟。在517nm下在bio-rad微板读取器中读取吸光度。将抗坏血酸和α-生育酚用作比较用已知抗氧化剂。dpph吸光度的减小即测量为自由基清除活性,并且使用以下等式来计算:抑制百分比=[o.d.对照物-o.d.经过处理/o.d.对照物]×100。
实施例10:(s,s)-sdg-1和(r,r)-sdg-2的还原能力
通过在fecl3存在下还原k3fecn6来测定合成的(s,s)-sdg-1、合成的(r,r)-sdg-2、天然的(s,s)-sdg-1、抗坏血酸和α-生育酚的还原能力,如通过所得三价铁-二价铁络合物的吸光度来测量(图2)。合成的(s,s)-sdg-1、合成的(r,r)-sdg-2和天然的(s,s)-sdg-1的还原能力在较高浓度下具有明显的浓度依赖性;然而,在所有测试浓度下,sdg都具有比已知的抗氧化剂抗坏血酸和α-生育酚相当或更高的还原能力,在200-500μm范围内效力有显著提高。在较低浓度(1-100μm)下观测到在还原能力与底物浓度之间有线性关系,从而允许为五种化合物建立回归线等式。这允许计算还原能力的一半最大有效浓度(ec50)(图3)。(s,s)-sdg-1和(r,r)-sdg-2的ec50(平均值±标准偏差)值分别是292.17±27.71μm和331.94±21.21μm。这些值与天然的(s,s)-sdg-1的值(ec50=275.24±13.15μm)相当,但是比抗坏血酸(ec50=1129.32±88.79μm)和α-生育酚(ec50=944.62±148.00μm)所显示的值高约三倍。
实施例11:清除羟基和过氧化氢自由基的能力
分别通过羟基自由基防止能力(horac,没食子酸标准物)和过氧化氢自由基吸光能力分析(orac,trolox标准物)来评估如表现为抑制荧光素氧化的(s,s)-sdg-1和(r,r)-sdg-2清除羟基和过氧化氢自由基的能力(表1)。合成的(s,s)-sdg-1和合成的(r,r)-sdg-2以浓度依赖性方式减小羟基自由基对荧光素的氧化并且发现比没食子酸高两倍。然而,可能由于痕量杂质,因此合成的(s,s)-sdg-1的活性不同于天然的(s,s)-sdg-1。在合成的(s,s)-sdg-1、(r,r)-sdg-2和天然的(s,s)-sdg-1存在下,使用2,2'-偶氮双(2-甲脒基丙烷)二盐酸盐(aaph)所产生的过氧化氢自由基对荧光素的氧化极大地减小,相对于trolox标准物效力提高两倍(表1)。
表1:合成和天然的sdg的抗氧化能力
a通过horac分析来测定;b通过orac分析来测定;c没食子酸当量;dtrolox当量。
在表1中,通过fenton反应从过氧化氢产生羟基自由基。由aaph(2,2'-偶氮双(2-甲脒基丙烷)二盐酸盐)产生过氧化氢自由基。测量荧光素的氧化。计算使用符合校准曲线的线性部分的sdg浓度。将结果呈现为平均值±标准偏差(n=3)。
实施例12:(s,s)-sdg-1和(r,r)-sdg-2的自由基清除活性
使用2,2-二苯基-1-苦味基偕腙肼(dpph)自由基清除分析来测定合成的(s,s)-sdg-1和(r,r)-sdg-2的自由基清除活性,并且与天然的(s,s)-sdg-1、抗坏血酸和α-生育酚的自由基清除活性相比(图4)。在低(5-25μm)和中浓度(50-100μm)范围下,sdg显示类似的清除潜力;然而在较高浓度(250-500μm)下,由合成的(s,s)-sdg-1所引起的抑制明显低于由(r,r)-sdg-2和天然的(s,s)-sdg-1所引起的抑制。为在低浓度和中浓度范围(5-100μm)下的潜力建立回归线允许测定这些化合物的自由基ec50清除活性。如图5中所示,合成的(r,r)-sdg-2(123.63±8.67μm)与合成的(s,s)-sdg-1(157.54±21.3μm)并非明显不同。这些值类似于天然的(s,s)-sdg-1(83.94±2.80μm)和α-生育酚(132.81±12.57μm)所显示的值,但是大大地低于抗坏血酸所示的值(439.56±11.81μm)。这些结果与对sdg所报道的结果相当(moree,s.;khanum,s.a.;rajesha,j.freerad.antiox.2011,1,31)。
本领域技术人员应了解在不脱离本发明的广泛发明构思的情况下,可对上述实施方案进行改变。因此,应理解本发明不限于所公开的具体实施方案,但意图涵盖在如由随附权利要求书所定义的本发明的精神和范围内的修改。
- 还没有人留言评论。精彩留言会获得点赞!