基于CRISPR/Cas技术敲除CAPNS1基因的人神经母细胞瘤细胞系的构建方法与流程
本发明涉及基因工程技术领域,尤其涉及敲除人神经母细胞瘤细胞capns1基因的方法。
背景技术
钙蛋白酶(calpain)是一组高度保守的特异性ca2+依赖性细胞内中性蛋白酶,其活性主要与细胞内ca2+浓度有关。calpain在机体的生理过程中发挥着重要作用,其不仅与细胞内蛋白质的水解关,还参与细胞自噬、细胞周期调控与凋亡、细胞骨架重构、葡萄糖转运、细胞信号转导等正常的生理过程[3,4]。自在大鼠的脑组织中首次发现一种可溶的ca2+依赖的中性蛋白酶以来,研究者们越来越关注这种蛋白酶在体内的作用。目前,研究发现在哺乳动物中calpain存在15种亚型,其中,calpain1(μ-calpain)和calpain2(m-calpain)在体内广泛表达,且研究也较为广泛。calpain1和calpain2主要由80kd的大亚基和30kd的小亚基(calpain-s1,capns1)组成,capns1是calpain1和calpain2活性所必需的,而且也是细胞迁移,凋亡和存活所必需的。
人神经母细胞瘤细胞(humanneuroblastomacell,hnc)是研究神经系统基因功能和神经毒性机制的良好的体外模型,建立敲除capns1基因的人神经母细胞瘤细胞模型有利于对capns1蛋白的作用的进一步研究,更有利于对神经系统多种疾病机理的研究。
基因敲除是通过dna序列改变,出现基因功能的“全或无”表象。基因沉默技术是以不改变dna序列为前提,使基因沉默,即基因不表达或低表达。基因编辑技术是指在基因组水平精确地进行基因编辑,其是通过精确识别靶细胞dna片段中靶点的核苷酸序列,利用核酸内切酶对dna靶点序列进行切割,从而完成对靶细胞dna目的基因片段的敲除、加入等过程。基因敲除构建出的突变型细胞系比基因沉默构建的细胞系稳定。细胞基因沉默的效率并不总是非常高,也不十分稳定,影响实验结果敏感性和可靠性。
继锌指核酸酶(zfn)和转录激活因子样效应物核酸酶(talens)基因编辑技术之后,第三代基因编辑技术crispr/cas技术是目前运用最为广泛的基因技术。crispr/cas是一种来源于原核生物适应性免疫系统,通过人工改良而得。相比zfn和talens,crispr/cas技术具有成本低、可同时进行多位点编辑、效率高等优势。crispr/cas系统分为3种,其中ii型最为简单,研究得也较多。该系统主要包含非特异性cas9核酸酶和sgrna两部分,其中sgrna具引导和检测作用;cas9是一种rna依赖的dna内切核酸酶,其利用单一向导rna(sgrna)使双链dna断裂,起到剪刀作用。
脱靶效应是存在于所有基因组靶向修饰技术中的一道难题,它会对基因组非特性序列进行切割,造成未知突变,增加后期的鉴定工作量。crispr/cas9系统中,对靶序列的识别主要是依靠一段20bp的短rna,但是研究表明当存在单个甚至多达5个碱基错配时,切割仍能正常发生。进一步研究发现,在这20bp中只有位于pam位点前12bp的种子序列对靶位点识别影响较大,即总共只有14bp(pam中的gg和种子序列)是靶位点识别的关键序列。这在生物体庞大的基因组中很容易出现脱靶位点,从而引入意外突变。除此之外,cas9蛋白或sgrna的浓度也对脱靶活性产生影响。
而神经细胞本身较其他细胞的培养较为苛刻,是一种转染质粒较难的细胞系,且转染效率低,而电转对神经细胞的伤害也较大,因此,对于转染条件的摸索较不易。在筛选单克隆时,神经细胞生长培养较难。综上,构建敲除人神经母细胞瘤细胞capns1基因并不容易。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于以crispr/cas技术敲除人神经母细胞瘤细胞capns1基因的方法及细胞系,其能够成功、稳定的敲除掉人神经母细胞瘤细胞中的capns1基因。
本发明提供了人capns1基因第4外显子和/或第5外显子在构建capns1基因敲除人神经母细胞瘤细胞中的应用。
本发明还提供了靶向capns1基因第4外显子的sgrna,其核苷酸序列如seqidno.1和seqidno.2所示。
靶向capns1基因第5外显子的sgrna,其核苷酸序列如seqidno.3所示。
本发明试验表明,采用capns1基因的第1~3外显子区域的靶位点构建出来的载体在转染细胞后,没有筛选到纯合子。而采用capns1基因的第4~5外显子的cds区作为靶点,成功地构建了capns1基因敲除sk-n-sh细胞株。
本发明还提供了如seqidno.4~9所示的核苷酸序列的6条敲除capns1基因的靶标寡核苷酸序列。
所述seqidno.4~9的核苷酸序列中,seqidno.4~5以seqidno.1为靶序列。seqidno.6~7以seqidno.2为靶序列。seqidno.8~9以seqidno.3为靶序列。
本发明还提供了敲除capns1基因的载体,包括骨架载体和dsdna片段;所述dsdna片段由seqidno.4~5退火形成;或由seqidno.6~7退火形成;或由seqidno.8~9退火形成。
本发明中,所述骨架载体为pgk1.1。
所述敲除capns1基因的载体的制备方法为,将dsdna与线性化的骨架载体t4dnaligase酶连接。
本发明还提供了敲除capns1基因的试剂,包括本发明提供的靶标寡核苷酸序列或本发明提供的敲除载体。本发明还提供了一种敲除人神经母细胞瘤细胞capns1基因的方法,将本发明提供的敲除载体转染入人神经母细胞瘤细胞中,获得敲除capns1基因的人神经母细胞瘤细胞株。
本发明中,所述人神经母细胞瘤细胞为sk-n-sh细胞。
所述sk-n-sh细胞为对数期的sk-n-sh细胞。
本发明中,所述转染采用电转染,转染的电压为1400v。
转染中,所述载体的浓度不低于1μg/μl。
以本发明提供方法构建的敲除capns1基因的人神经母细胞瘤细胞。
本发明提供了敲除人神经母细胞瘤细胞capns1基因的方法,质粒转染sk-n-sh细胞后,制备单克隆,cruisertmenzyme酶切后疑似为阳性克隆,单克隆测序结果显示sk-n-sh细胞capns1基因敲除细胞系构建成功;并且,在capns1-/-组,capns1蛋白及calpain1和calpain2蛋白水平明显降低。结果表明sk-n-sh细胞capns1基因敲除细胞系构建成功。
附图说明
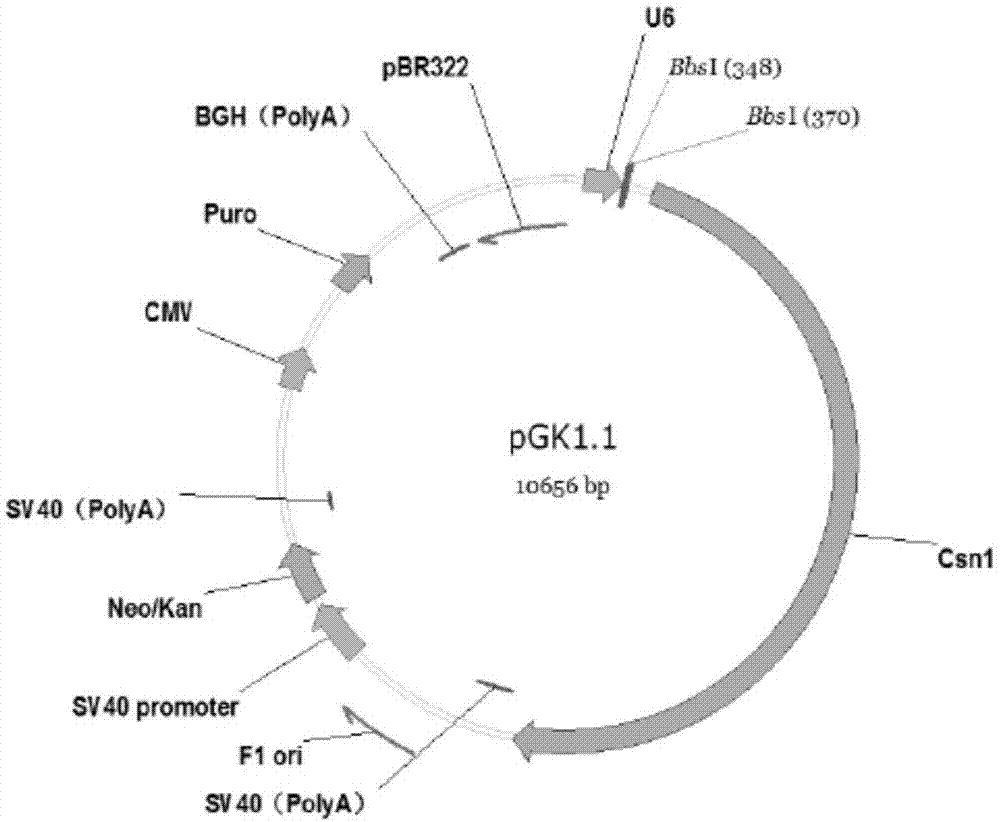
图1pgk1.1质粒图谱;
图2载体酶切后的电泳检测图;其中泳道1是原质粒对照,m为marker,2-4为切开的载体
图3菌落pcr检测电泳图;m为marke菌落pcr检测电泳图k;1、2为capns1-grna1菌落pcr检测电泳图;3、4为capns1-grna2菌落pcr检测电泳图;5、6为capns1-grna3菌落pcr检测电泳图
图4sgrna测序验证;a、b、c分别代表插入pgk1.1载体的sgrna1、sgrna2、sgrna3序列测序图;
图5sgrna测序比对结果;a、b、c分别表示sgrna1位点、sgrna2位点、sgrna3位点比对结果;
图6capns1基因cruiser验证;
图7sk-n-sh细胞capns1基因敲除细胞系单克隆测序;
图8capns1基因敲除sk-n-sh细胞ta克隆测序及比对;
图9capns1敲除后capns1、calpain1和calpain2蛋白表达情况;
图10不同电压转染sk-n-sh细胞结果;
图11capns1基因敲除载体转染后的混克隆测序验证。
具体实施方式
本发明提供了以crispr/cas技术敲除人神经母细胞瘤细胞capns1基因的方法及细胞系,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
本发明采用的试材皆为普通市售品,皆可于市场购得。
下面结合实施例,进一步阐述本发明:
实施例
1)设计crispr/cas9敲除靶位点
首先,需要在靶标dna区域中设计一对20bp左右的oligodna,通过以下在线工具设计:
麻省理工学院的crisprdesign:http://crispr.mit.edu/
选取capns1基因所有转录本公共的cds区,找出公共cds区所处的外显子进行靶位点设计,选择第四个和第五个外显子进行靶位点设计。
3个靶位点序列信息:
capns1-grna1:agttcgacactgaccgatcaggg(seqidno.1)
capns1-grna2:tgaactcccaggtgcctttgagg(seqidno.2)
capns1-grna3:cactttcatctgagtagcgtcgg(seqidno.3)
2)引物添加接头
引物合成需在靶序列头部添加额外的碱基,正向引物添加cacc,反向引物添加aaac,需要特别注意的是靶序列的第一个碱基必须是g,如果你选取的靶序列第一个碱基不是g,可自行在靶序列前加一个g,靶序列引物设计如下:
capns1-1f:caccgagttcgacactgaccgatca(seqidno.4)
capns1-1r:aaactgatcggtcagtgtcgaactc(seqidno.5)
capns1-2f:caccgtgaactcccaggtgcctttg(seqidno.6)
capns1-2r:aaaccaaaggcacctgggagttcac(seqidno.7)
capns1-3f:caccgcactttcatctgagtagcgt(seqidno.8)
capns1-3r:aaacacgctactcagatgaaagtgc(seqidno.9)
3)oligo杂交,敲除载体连接反应
将合成后的2条单链oligodna稀释成10μm,退火形成dsdna,再与线性化后的pgk1.1linearvector(cat.no.gp0134,图谱如图1)载体连接,可直接用t4dnaligase连接,退火反应体系如下:
将以上体系瞬时离心后,置于pcr仪中95℃孵育3min,孵育后自然冷却20min。取1μl的杂交后的dsdna进行t4dnaligase连接反应,反应体系如下:
将以上体系瞬时离心后,置于pcr仪中16℃孵育30min。
pgk1.1质粒包含了含cas9核酸酶表达框和grna克隆框2个重要原件,其图谱如下,大小为10656bp。vspprimer:catatgcttaccgtaactt
gaaag序列位于载体的u6启动子区域。下游负链oligo序列即靶位点的反向互补序列,位于载体的双bbsi酶切位点。(靶位点替换双bbsi酶切位点)。载体经酶切后,获得10kb左右片段,见图2。
4)转化top10感受态
①从-80℃冰箱中取1管top10感受态,置冰上融解。
②融解后加入10μl连接产物,轻弹混匀,冰上孵育30min。
③42℃水浴热击60sec,迅速拿出置冰上冷却2~3min。
④向管中加入800μl无抗soc液体培养基,至摇床(37℃/160rpm)恢复培养培养45min。
⑤4500rpm离心5min,弃去800μl上清,将沉淀悬在剩余的100μl上清中,均匀涂布于含kan抗性的的筛选平板上,倒置培养过夜。
5)筛选阳性重组子
第二天使用上游引物vspprimer与下游负链oligo引物进行菌落pcr筛选,阳性克隆pcr正确大小应为100bp,筛选到的阳性克隆抽质粒进一步测序验证。测序正确的质粒浓缩到1μg/μl浓度以上。
使用上游引物vspprimer与下游负链oligo引物进行菌落pcr筛选,均能扩增出100bp大小的片段,说明所检测单克隆全为阳性,见图3。测序发现插入序列正确,见图4。seqman软件测序比对结果正确,载体构建成功,见图5。
6)电转染靶细胞(cat.no.gp7901)
①取状态良好的对数生长期sk-n-sh细胞悬液台盼蓝计数,确定细胞数及细胞活力(细胞活力>95%)。
②取5×106个细胞于15ml离心管中,离心弃上清(1000rpm/4min)。
③将细胞沉淀悬浮于210μldpbs中,转移至1.5mlep管中,加入所需量构建好的敲除质粒5~8μg(质粒浓度要求1μg/μl以上),轻轻混匀。
④将上述细胞质粒混合液用专用电转枪头转移至电击杯中,确定电击杯中溶液无气泡,且液面凸起后,盖上电击杯盖并至于电转仪,设定好电转条件后进行电转。待显示峰图正常后取出细胞液转移至六孔板培养基中(培养基需事先37℃预热且无抗生素)。
结论:1200v、1300v和1400v三个条件,1400v电转效率较高且死细胞较少,建议使用1400v。
药杀浓度结果:puro1ug/ml;
单克隆验证结果:能长成单克隆细胞,3cell/孔;图10。
7)pool细胞测序检测敲除效率
①电转72hr后,pool细胞台盼蓝计数。
②在筛选阳性克隆之前,需对pool细胞(混合克隆)的敲除效率进行体内验证。
③一般靶位点附近的序列测序中(图11),阳性样品应在靶点位置及之后的序列中出现套峰,如敲除效率较低时,信号强度往往较低,影响判断;
根据图11的测序结果,可以看出在靶点位置及之后的序列中明显出现套峰,初步认为阳性克隆的存在。
8)单克隆的制备和生长
有限稀释法稀释细胞至10块96孔板中,37℃,co2培养箱中静置培养;一周后观察单克隆生长情况,约两周后将长起来的单克隆转移至48孔中扩大培养;当细胞长满48孔1/2时,即可取出一部分(102~104),使用genlocitna抽提试剂盒(cat.no.gp0155,gp0156)提取细胞基因组。
9)单克隆基因组dna的提取
取102~104个细胞于1.5mlep管中,室温1500rpm离心5min,小心吸掉培养液。
加入150μlpbs重悬细胞,室温1500rpm离心5min,小心弃去上清。
重复步骤2一次。
向离心管中加入适量体积(推荐体积为50~200μl)预先配制的溶液a与溶液b的混合液,枪头吹打5次,冰上放置10min,使细胞充分裂解。
加入两倍体积的无水乙醇,颠倒混匀,-20℃条件下沉淀20min以上。
4℃,12000rpm,离心20min,弃上清。
加入400~500μl预冷的75%乙醇洗涤沉淀,4℃,12000rpm,离心10min,小心弃去上清,自然晾干(不超过5min为宜)。
加入适量体积(推荐体积为10-30μl)灭菌的双蒸水溶解沉淀,溶液可直接用于pcr反应,或于-20℃保存。
10)pcr扩增目的片段
引物设计
在敲除靶位点附近设计高特异性的primers,扩增产物长度为分别为455bp。primers引物序列如下:
capns1-seqf3:aagccatggagacactatgc
capns1-seqr3:tggtccatagaggtcatagg
pcr扩增获得杂交dna
在灭菌pcr管中配制如下反应体系,使用高特异性的primers,扩增获得野生型和突变型充分杂交的dna产物。
pcr反应程序如下:
自然冷却至40℃以下(野生型片段与突变型片段杂交)。
pcr结束后,取2~3μl进行电泳检测,要求目的片段明亮并且单一。
11)cruiserrenzyme酶切筛选阳性克隆
在灭菌pcr管中配制如下反应体系:
45℃反应20min后立即向上述10μl反应体系内加入2μl6×stopbuffer,随后进行琼脂糖电泳检测或置于-20℃保存。
根据扩增引物及理论敲除位点位置,扩增产物约455bp,扩增目的大小明显变小或者可以切出条带的克隆为疑似阳性克隆。从图6可以看出,呈现的条带在400bp和500bp之间,疑似为阳性克隆。
12)测序筛选阳性克隆
对crusier酶切初步筛选出来的阳性克隆进行测序验证,进一步确认阳性克隆(图7)。ta克隆测序结果比对发现两个亲本都缺失35bp:tcagggaccatttgcagtagtgaactcccaggtgc,见图8。capns1基因敲除sk-n-sh细胞株构建成功。
13)ta克隆
对于阳性克隆中,两个等位基因突变情况不一样的阳性克隆,重新做ta克隆后送测序,跟野生型比对,确定每个等位基因的突变情况。
14)westernblot检测capns1、calpain1和calpain2蛋白表达情况
对稳定capns1基因敲除后的sk-n-sh细胞中的capns1蛋白及活性受capns1调节的calpain1和calpain2蛋白表达进行westernblot分析,结果显示,与对照sk-n-sh细胞(capns1+/+)相比,稳定capns1基因敲除后的sk-n-sh细胞(capns1-/-)中capns1、calpain1和calpain2的蛋白表达明显降低。见图9。
以上仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
序列表
<110>南华大学
<120>基于crispr/cas技术敲除capns1基因的人神经母细胞瘤细胞系的构建方法
<130>mp1803791
<160>9
<170>siposequencelisting1.0
<210>1
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>1
agttcgacactgaccgatcaggg23
<210>2
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>2
tgaactcccaggtgcctttgagg23
<210>3
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>3
cactttcatctgagtagcgtcgg23
<210>4
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>4
caccgagttcgacactgaccgatca25
<210>5
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>5
aaactgatcggtcagtgtcgaactc25
<210>6
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>6
caccgtgaactcccaggtgcctttg25
<210>7
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>7
aaaccaaaggcacctgggagttcac25
<210>8
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>8
caccgcactttcatctgagtagcgt25
<210>9
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>9
aaacacgctactcagatgaaagtgc25
- 还没有人留言评论。精彩留言会获得点赞!