一种噁嗪苯醚衍生物及其制备方法与流程
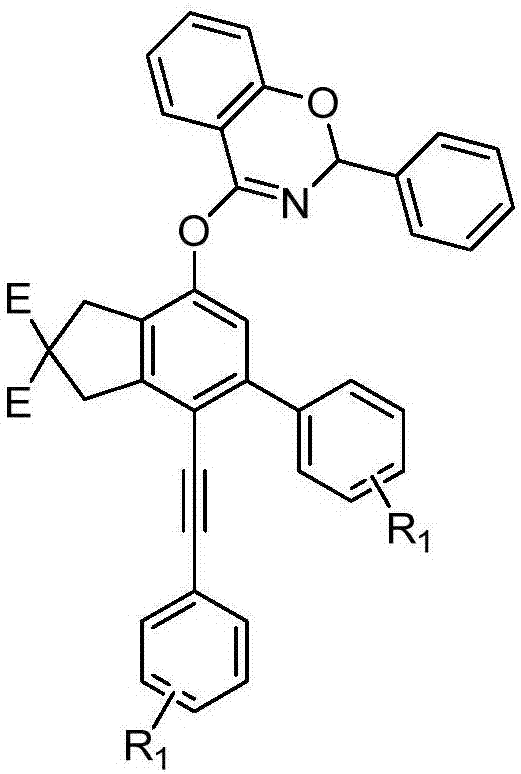
本发明属于有机化合物领域,具体涉及一种噁嗪苯醚衍生物及其制备方法。
背景技术
噁嗪苯醚衍生物有多环的存在,其结构更加复杂多样。噁嗪苯醚衍生物优良的耐热性,被广泛使用在高耐热覆铜板上,在航空航天,汽车火车等诸多领域也得到广泛应用;也被用于香料工业、合成树脂和其他有机合成生产;噁嗪苯醚衍生物种类众多,部分噁嗪苯醚衍生物具有医药价值,受到国内外众多医药企业和研究机构的重视,近几年来,不断有噁嗪苯醚医药衍生物问世;噁嗪苯醚衍生物在化工生产中也将表现出更加广阔的用途前景。
技术实现要素:
本发明的目的在于提供一种噁嗪苯醚衍生物,具有多环存在,结构更复杂,有广阔的应用前景。
本发明的目的在于提供一种噁嗪苯醚衍生物的制备方法,简便、反应时间短,效率高。
本发明具体技术方案如下:
本发明提供的一种噁嗪苯醚衍生物,结构式为:
其中e=co2r,r为直链烷基、支链烷基或不饱和烃基;r1为氢、卤素、直链烷基、支链烷基、酯基、烷氧基或其相应的衍生物。
进一步的,所述r为甲基,r1为甲基,其结构式为以下式ⅰ;所述r为乙基,r1为乙基,其结构式为以下式ⅱ;所述r为异丙基,r1为氢,其结构是为以下式ⅲ:
上述噁嗪苯醚衍生物的制备方法为:
加热条件下,将四炔在甲苯溶剂中与2-苯基-3,4-二氢-2h-苯并[1,3]噁嗪-4-酮反应,自然冷却至室温,停止反应;将产物纯化分离,得到白色固体,即噁嗪苯醚衍生物。
进一步的,四炔、2-苯基-3,4-二氢-2h-苯并[1,3]噁嗪-4-酮与甲苯的摩尔比的摩尔比为1:1:28-66;
所述四炔的结构式为:
进一步的,所述加热条件下是指加热至110-115℃的条件下,所述反应是指反应15小时以上。
所述纯化分离具体为:将所得产物用水洗涤,乙酸乙酯萃取,减压旋干,用体积比1:20的乙酸乙酯:石油醚的柱层析分离,得到白色固体,即噁嗪苯醚衍生物。柱层析产率约为75.6%。
所述的四炔的制备方法为:
1)以氢化钠为催化剂,将丙二酸酯与炔丙基溴加入到无水乙腈中冰水浴,反应,然后纯化分离,得到黄棕色固体产物,即化合物1;
2)将化合物1与苯乙炔基溴或取代的苯乙炔基溴混合在pd(pph3)2cl2/cui的无水无氧催化体系中,以三乙胺作碱,以无水乙腈为溶剂,室温下搅拌反应,纯化分离后,得到浅黄色固体产物,即前体化合物2,即四炔;
进一步的,步骤1)中氢化钠、丙二酸酯、炔丙基溴与无水乙腈的摩尔比为4-5:1:2.2-3.2:20-23;所述丙二酸酯选自丙二酸二甲酯、丙二酸二乙酯或丙二酸二异丙酯。
步骤1)的反应具体为:温度在0-5℃,反应时间在8小时以上;
步骤1)中所述纯化分离具体为:产物加水洗涤,用乙酸乙酯萃取,减压旋干,得到黄棕色固体产物,即化合物1。
步骤1)中所述化合物1结构式为
步骤2)中所述化合物1与苯乙炔基溴或取代的苯乙炔基溴、pd(pph3)2cl2/cui、三乙胺和无水乙腈的物质的量比为1:2.2-3.2:0.03-0.04:4-5:30-45;
步骤2)所述搅拌反应,时间在12小时以上;所述取代的苯乙炔基溴选自对甲基苯乙炔基溴或对乙基苯乙炔基溴;
步骤2)中所述纯化分离具体为:产物用水洗涤,用乙酸乙酯萃取,减压旋干,用体积比为1:40的乙酸乙酯:石油醚柱层析分离,得到浅黄色固体产物,即前体化合物2,即四炔。
步骤2)中所述pd(pph3)2cl2/cui的无水无氧催化体系中,摩尔比pd(pph3)2cl2:cui=3:1。
本发明在无催化剂、无添加剂、无保护的状态下,以甲苯为溶剂,四炔与2-苯基-3,4-二氢-2h-苯并[1,3]噁嗪-4-酮在110-115℃的条件下反应15h以上,反应过程首先由四炔底物自身环化,由于在于四炔生成的苯炔是缺电子的中间体,极不稳定,具有高度活泼性。四炔经过hdda反应生成苯炔中间体,然后高度活泼的苯炔中间体与2-苯基-3,4-二氢-2h-苯并[1,3]噁嗪-4-酮发生亲核加成,氮原子上的氢正离子迁移到苯炔中间体的碳负离子上,从而得到噁嗪苯醚衍生物。
与现有技术相比,本发明提供了一系列新的噁嗪苯醚衍生物。相对于普通噁嗪苯醚衍生物,本发明制备的噁嗪苯醚衍生物有多环的存在,其结构更加复杂多样,噁嗪苯醚衍生物优良的耐热性,被用于香料工业、合成树脂和其他有机合成生产,部分噁嗪苯醚衍生物具有医药价值,在化工生产中也将表现出更加广阔的用途前景。并且,本发明提供的制备方法简便、反应时间短,效率高。
附图说明
图1为噁嗪苯醚衍生物的结构式;e=co2r,r为直链烷基、支链烷基或不饱和烃基;r1为卤素、直链烷基、支链烷基、酯基、烷氧基或其相应的衍生物;
图2为实施例1制备的噁嗪苯醚衍生物的结构式;
图3为实施例1制备的噁嗪苯醚衍生物的核磁共振氢谱;
图4为实施例1制备的噁嗪苯醚衍生物的核磁共振碳谱;
图5为实施例2制备的噁嗪苯醚衍生物的结构式;
图6为实施例2制备的噁嗪苯醚衍生物的核磁共振氢谱;
图7为实施例2制备的噁嗪苯醚菲衍生物的核磁共振碳谱;
图8为实施例3制备的噁嗪苯醚衍生物的结构式;
图9为实施例3制备的噁嗪苯醚衍生物的核磁共振氢谱;
图10为实施例3制备的噁嗪苯醚衍生物的核磁共振碳谱;
图11为实施例1的反应机理示意图。
具体实施方式
一种噁嗪苯醚衍生物,所述的噁嗪苯醚衍生物结构式为:
一种噁嗪苯醚衍生物的制备方法,所述的制备方法包括以下步骤:
(1)以830mmol氢化钠为催化剂,将200mmol丙二酸二甲酯与440mmol炔丙基溴加入到210ml无水乙腈中冰水浴,搅拌反应8小时,产物加水洗涤,用乙酸乙酯萃取,减压旋干,得到棕黄色固体产物,即化合物1;
(2)将80mmol化合物1与200mmol对甲基苯乙炔基溴混合在pd(pph3)2cl2/cui的无水无氧催化体系中(2.56mmol/0.85mmol),摩尔比pd(pph3)2cl2:cui=3:1,以336mmol三乙胺作碱,以150ml无水乙腈为溶剂,室温下搅拌反应12小时,产物用水洗涤,用乙酸乙酯萃取,减压旋干,用体积比为1:40的乙酸乙酯:石油醚柱层析分离,得到浅黄色固体产物,即前体化合物2,即四炔。
(3)在110℃的条件下,步骤(2)所制备的1mmol四炔在5ml甲苯溶剂与1mmol2-苯基-3,4-二氢-2h-苯并[1,3]噁嗪-4-酮反应15小时,得化合物3,即噁嗪苯醚衍生物的粗产物;将制备的噁嗪苯醚衍生物的粗产物用水洗涤,乙酸乙酯萃取,减压旋干,用体积比乙酸乙酯:石油醚=1:20柱层析分离,得到白色固体产物,即噁嗪苯醚衍生物,柱层析产率约为78.6%。
白色固体产物结构通过;1hnmr;13cnmr来测定。
白色固体产物:
1hnmr(500mhz,cdcl3)δ7.59-7.58(m,1h),7.52(m,2h)7.51(m,2h),7.35-7.33(m,1h),7.29(m,3h),7.27-7.26(m,5h),7.26-7.23(m,2h),7.12(s,1h),7.11(s,1h),6.61(s,1h),3.88(s.2h),3.87-3.74(m,6h),3.68(s,2h),2.41(s,3h),2.35(s,3h)。
13cnmr(125mhz,cdcl3)δ172.26,157.69,157.18,148.32,145.97,144.47,140.07,138.75,137.72,137.36,131.72,131.50,129.62,129.47,129.05,128.76,127.40,125.36,122.41,121.84,116.88,96.27。
实施例2
一种噁嗪苯醚衍生物,所述的噁嗪苯醚衍生物结构式为:
一种噁嗪苯醚衍生物的制备方法,所述的制备方法包括以下步骤:
(1)以氢830mmol化钠为催化剂,将200mmol丙二酸二乙酯与440mmol炔丙基溴加入到210ml无水乙腈中冰水浴,搅拌反应8小时,产物加水洗涤,用乙酸乙酯萃取,减压旋干,得到棕黄色固体产物,即化合物1;
(2)将80mmol化合物1与200mmol对乙基苯乙炔基溴混合在pd(pph3)2cl2/cui的无水无氧催化体系中(2.56mmol/0.85mmol),摩尔比pd(pph3)2cl2:cui=3:1,以336mmol三乙胺作碱,150ml无水乙腈为溶剂,室温下搅拌反应12小时,产物用水洗涤,用乙酸乙酯萃取,减压旋干,用体积比为1:40的乙酸乙酯:石油醚柱层析分离,得到浅黄色固体产物,即前体化合物2,即四炔。
(3)在110℃的条件下,步骤(2)所制备的1mmol四炔在5ml甲苯溶剂与1mmol2-苯基-3,4-二氢-2h-苯并[1,3]噁嗪-4-酮反应15小时,得噁嗪苯醚衍生物的粗产物;将制备的噁嗪苯醚衍生物的粗产物用水洗涤,乙酸乙酯萃取,减压旋干,用体积比乙酸乙酯:石油醚=1:20柱层析分离,得到白色固体产物,即噁嗪苯醚衍生物,柱层析产率约为79.5%。
白色固体产物结构通过;1hnmr;13cnmr来测定。
白色固体产物:
1hnmr(500mhz,cdcl3)δ7.62-7.61(m,1h),7.53(m,2h),7.52(m,2h),7.35(s,1h),7.34-7.30(m,8h),7.29-7.28(m,2h),7.26-7.24(m,1h),7.15-7.14(m,1h),6.62(s,1h),4.22(q,j=10.0hz,4h),3.88(s,2h),3.69(s.2h),2.73(q,j=5.0hz,2h),2.64(q,j=10.0hz,2h),1.30(t,j=10.0hz,6h),1.24(s,6h);
13cnmr(125mhz,cdcl3)δ171.81,163.85,157.74,157.15,148.29.147.08,146.07,144.99,143.97,140.04,137.64,134.79,131.79,129.68,128.94,128.23,127.78,125.35,122.33,121.79,116.85,113.72,96.24,89.56,86.83,62.29,60.01,41.73,38.90,29.25,29.05,15.99,15.81,14.44。
实施例3
一种噁嗪苯醚衍生物,所述的噁嗪苯醚衍生物结构式为:
一种噁嗪苯醚衍生物的制备方法,所述的制备方法包括以下步骤:
(1)以氢830mmol化钠为催化剂,将200mmol丙二酸二异丙酯与440mmol炔丙基溴加入到210ml无水乙腈中冰水浴,搅拌反应8小时,产物加水洗涤,用乙酸乙酯萃取,减压旋干,得到棕黄色固体产物,即化合物1;
(2)将80mmol化合物1与200mmol苯乙炔基溴混合在pd(pph3)2cl2/cui的无水无氧催化体系中(2.56mmol/0.85mmol),摩尔比pd(pph3)2cl2:cui=3:1,以336mmol三乙胺作碱,150ml无水乙腈为溶剂,室温下搅拌反应12小时,产物用水洗涤,用乙酸乙酯萃取,减压旋干,用体积比为1:40的乙酸乙酯:石油醚柱层析分离,得到浅黄色固体产物,即前体化合物2,即四炔。
(3)在110℃的条件下,步骤(2)所制备的1mmol四炔在5ml甲苯溶剂与1mmol2-苯基-3,4-二氢-2h-苯并[1,3]噁嗪-4-酮反应15小时,得化合物5,即噁嗪苯醚衍生物的粗产物;将制备的噁嗪苯醚衍生物的粗产物用水洗涤,乙酸乙酯萃取,减压旋干,用体积比乙酸乙酯:石油醚=1:20柱层析分离,得到白色固体产物,即噁嗪苯醚衍生物,柱层析产率约为76.7%。
白色固体产物结构通过;1hnmr;13cnmr来测定。
白色固体产物:
1hnmr(500mhz,cdcl3)δ7.68-7.67(m,1h),7.52-7.51(m,2h),7.43-7.42(m,2h),7.36(s,3h),7.34-7.31(m,8h),7.30-7.29(m,2h),7.26-7.24(m,1h),6.98(s,1h),6.61(s.1h),5.09-5.04(m,2h),3.85(s,2h),3.66(s,2h),1.26-1.22(m,12h)。
13cnmr(125mhz,cdcl3)δ173.61,173.28,171.32,157.78,157.18,154.29,152.12,148.47,146.34.141.69,140.17,140.01,134.82,131.78,129.78,128.66,128.28,127.38,125.35,123.93,122.44,121.83,116.87,115.60,113.74,111.76,95.98,95.81,89.53,87.31,69.82,60.07,41.65,38.80,21.95。
- 还没有人留言评论。精彩留言会获得点赞!
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 吡唑衍生物及其作为大麻素受体介体的用途的制造方法与工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 水生态处理中淤泥及衍生物的再利用处理系统的制造方法与工艺
- Schiglautone A衍生物的组合物用于制备抗缺氧药物的制作方法
- Schiglautone A衍生物的组合物用于制备抗红细胞低下性贫血药物的制作方法
- 用于生产化学品及其衍生物的组合物和方法与制造工艺
- 7‑OH‑8‑[(1‑胺基‑1‑苯基衍生物)‑次甲基]‑黄酮化合物及其制备方法及其应用与制造工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 含D型非天然氨基酸的抗菌肽类似物及其合成与应用的制造方法与工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 一种白杨素氨基酸衍生物的制备方法与制造工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 水生态处理中淤泥及衍生物的再利用处理系统的制造方法与工艺
- 一种新型罗汉果醇衍生物单体的制作方法
- Schiglautone A衍生物的组合物用于制备抗鼻炎药物的制作方法
- Schiglautone A衍生物的组合物用于制备防治肾纤维化药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗心衰药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Schiglautone A 衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备防治胰腺纤维化药物的制作方法