一种咪唑并异吲哚类衍生物游离碱的晶型及其制备方法与流程
本发明涉及(s)-2-(4-(4-(4-(6-氟-5h-咪唑并[5,1-a]异吲哚-5-基)哌啶-1-基)苯基)-1h-吡唑-1-基)乙醇的ii晶型及其制备方法,其在药物组合物中的应用以及该ii晶型、组合物在制备治疗具有ido介导的色氨酸代谢途径病理学特征的疾病中的用途。
背景技术:
肿瘤生物治疗是应用现代生物技术及其相关产品进行肿瘤防治的新疗法,因其安全、有效、不良反应低等特点,成为继手术、放疗、化疗之后肿瘤治疗的第四种模式,其通过调动宿主的天然防御机制(比如抑制ido介导的肿瘤免疫逃逸机制)或给予天然产生的靶向性很强的物质来获得抗肿瘤的效应。
吲哚胺-吡咯-2,3-双加氧酶(indoleamine-pyrrole-2,3-dioxygenase,ido)是一种含铁血红素单体蛋白,由403个氨基酸残基组成,包括两个折叠的α-螺旋结构域,大结构域包含催化口袋,底物可在催化口袋内与ido发生疏水等作用。ido是催化色氨酸转化为甲酰犬尿氨酸的酶,广泛分布在人和其他哺乳动物(兔、鼠)除肝脏以外的组织中,是肝脏以外唯一可催化色氨酸分解代谢的限速酶,而色氨酸是细胞维持活化和增殖所必需的氨基酸,也是构成蛋白质不可缺少的重要成分。ido与干扰素(interferon,ifn)、白细胞介素(interleukin,il)、肿瘤坏死因子等多种细胞因子关系密切,它们在一定条件下可激活ido。而t-细胞的细胞周期中存在一个对色氨酸水平非常敏感的调节点,一方面,ido使局部色氨酸耗竭,致使t-细胞停滞于g1期中期,从而抑制了t细胞的增殖;另一方面,ido催化色氨酸代谢产生的主要产物犬尿素由氧自由基介导引起细胞内氧化剂和抗氧化剂改变而诱导t-细胞凋亡,这是存在于机体的固有的免疫抑制机制。目前大量研究表明ido在白血病细胞中较高表达,使局部t细胞增殖受抑,抑制t-细胞介导的免疫反应,使t-细胞活化信号转导受阻,从而介导肿瘤细胞逃逸免疫系统的攻击。已经发现大多数人类肿瘤组成性地表达ido。因此,ido是一个具潜力的癌症免疫治疗的靶标。
公开的选择性抑制ido的抑制剂专利申请包括wo2012142237、wo2004094409、wo2006122150、wo2007075598、wo2010005958和wo2014066834等。
ido抑制剂作为药物在医药行业具有良好的应用前景,本申请人在专利申请wo2016169421中提供了一种结构新型的高效低毒的选择性ido抑制剂化合物,具有优异的效果和作用,特别是优异的药代吸收活性,其化学名为(s)-2-(4-(4-(4-(6-氟-5h-咪唑并[5,1-a]异吲哚-5-基)哌啶-1-基)苯基)-1h-吡唑-1-基)乙醇,结构如下所示
作为药用活性成分的晶型结构往往影响到该药物的化学稳定性,结晶形式、制备方法及储存条件的不同有可能导致化合物的晶型结构的变化,有时还会伴随着产生其他形态的晶型。一般来说,无定形的药物产品没有规则的晶体结构,往往具有其它缺陷,比如产物稳定性较差,过滤较难,易结块,流动性差等,这些差异往往导致生产放大时的困难。而现有晶型的稳定性有待提高。因此,改善化合物的各方面性质是很有必要的,我们需要深入研究找到晶型纯度较高并且具备良好化学稳定的新晶型。
技术实现要素:
本发明要解决的技术问题是提供一种咪唑并异吲哚类衍生物的游离碱(s)-2-(4-(4-(4-(6-氟-5h-咪唑并[5,1-a]异吲哚-5-基)哌啶-1-基)苯基)-1h-吡唑-1-基)乙醇的ii晶型,该晶型具备良好的晶型稳定性和化学稳定性,可更好地应用于临床。
本发明的技术方案如下:
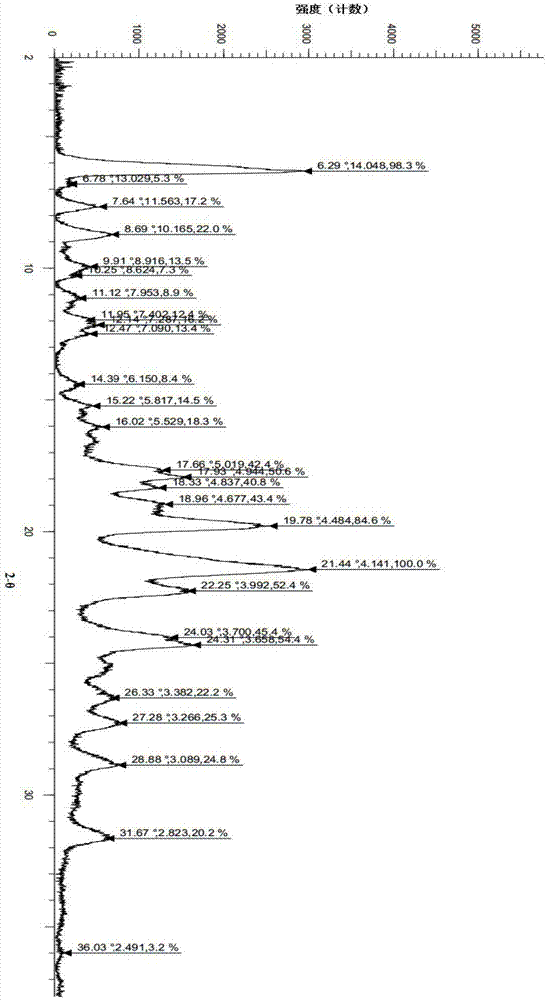
本发明提供一种式(i)所示化合物的ii晶型,其特征在于:使用cu-kα辐射,得到以衍射角2θ角度表示的x-射线粉末衍射图谱,其在衍射角2θ角6.3、7.6、8.7、9.9、11.1、12.1、14.4、15.2、17.9、19.8、21.4、24.3、26.3、27.3、28.9和31.6处有特征峰,且在20.8或21.1处没有特征峰,
在本发明的一个优选实施例方案中,本发明提供一种式(i)所示化合物的ii晶型,其特征在于:使用cu-kα辐射,得到以衍射角2θ角度表示的x-射线粉末衍射图谱,其在衍射角2θ角6.29、7.64、8.69、9.91、11.12、11.95、12.14、12.47、14.39、15.22、16.02、17.93、18.33、19.78、21.44、22.25、24.03、24.31、26.33、27.28、28.88和31.67处有特征峰,且在20.8和21.1处没有特征峰。
在本发明的一个优选实施例方案中,本发明提供一种式(i)所示化合物的ii晶型,其特征在于:使用cu-kα辐射,得到以衍射角2θ角度表示的x-射线粉末衍射图谱,其在衍射角2θ角6.29、6.78、7.64、8.69、9.91、10.25、11.12、11.95、12.14、12.47、14.39、15.22、16.02、17.66、17.93、18.33、18.96、19.78、21.44、22.25、24.03、24.31、26.33、27.28、28.88、31.67和36.03处有特征峰,且在20.8和21.1处没有特征峰。
在本发明的一个优选实施例方案中,本发明提供一种式(i)所示化合物的ii晶型,其特征在于:使用cu-kα辐射,得到的x-射线粉末衍射图谱如图1所示。
在本发明的一个优选实施例方案中,本发明提供一种式(i)所示化合物的ii晶型,其特征在于:熔点为210℃~218℃,优选为214℃~216℃。
在本发明的一个优选实施例方案中,本发明进一步提供一种制备式(i)所示化合物的ii晶型的方法,所述方法包括:
(1)方法一,将式(i)所示化合物溶解于适量的溶剂中,析晶,过滤结晶并洗涤,干燥后即可得到目标ii晶型,所述溶剂可以是醚类、卤代烷类、醇类溶剂,所述醚类溶剂优选四氢呋喃,乙二醇甲醚或1,4-二氧六环,所述卤代烷类溶剂优选二氯甲烷,所述醇类溶剂优选乙醇或甲醇;
(2)方法二,通过将式(i)所示化合物溶解于适量的良溶剂中,加入反溶剂,进行析晶,过滤结晶并洗涤,干燥后即可得到目标ii晶型,所述良溶剂可以为二甲基亚砜、甲醇、乙醇、丙酮中的一种或多种,所述反溶剂可以是水或脂肪烃;
本发明进一步涉及式(i)所示化合物的ii晶型的药物组合物,其特征在于包含一种或多种药学上可接受的载体、稀释剂或赋形剂。
本发明进一步涉及式(i)所示化合物的ii晶型或ii晶型的药物组合物在制备治疗具有ido介导的色氨酸代谢途径病理学特征的疾病的药物中的用途,所述疾病选自癌症、阿尔茨海默病、自身免疫性疾病、抑郁症、焦虑症、白内障、心理障碍或艾滋病;所述癌症选自乳腺癌、宫颈癌、结肠癌、肺癌、胃癌、直肠癌、胰腺癌、脑癌、皮肤癌、口腔癌、前列腺癌、骨癌、肾癌、卵巢癌、膀胱癌、肝癌、输卵管肿瘤、卵巢瘤、腹膜肿瘤、iv期黑色素瘤、神经胶质瘤、神经胶母细胞瘤、肝细胞癌、乳突肾性瘤、头颈部肿瘤、白血病、淋巴瘤、骨髓瘤或非小细胞肺癌。
通过x-射线粉末衍射图谱(xrpd)、差示扫描量热分析(dsc)对所得到式(i)所示化合物的ii晶型进行结构测定、晶型研究。
本发明中晶型的析晶方法是常规的,例如挥发析晶、降温析晶或室温下析晶。
本发明晶型制备方法中所用的起始原料可以是任意形式的式(i)所示化合物,具体形式包括但不限于:无定形、任意晶型等。
在本申请的说明书和权利要求书中,除非另有说明,否则本文中使用的科学和技术名词具有本领域技术人员所通常理解的含义。然而,为了更好地理解本发明,下面提供了部分相关术语的定义和解释。另外,当本申请所提供的术语的定义和解释与本领域技术人员所通常理解的含义不一致时,以本申请所提供的术语的定义和解释为准。
本发明所述的“打浆”是指利用物质在溶剂中溶解性差,但杂质在溶剂中溶解性好的特性进行纯化的方法,打浆提纯可以去色、改变晶型或去除少量杂质。
本发明所述“卤代”是指被“卤素原子”取代,“卤素原子”是指氟原子、氯原子、溴原子、碘原子等。
本发明所述的“醚类溶剂”是指含有醚键-o-且碳原子数为1至10个的链状化合物或环状化合物,具体实例包括但不限于:丙二醇甲醚、四氢呋喃或1,4-二氧六环。
本发明所述的“醇类溶剂”是指一个或多个“羟基”取代“c1-6烷基”上的一个或多个氢原子所衍生的基团,所述“羟基”和“c1-6烷基”如前文所定义,具体实例包括但不限于:甲醇、乙醇、丙醇或2-丙醇。
本发明所述的“良溶剂”是指对分子(或溶质)具有较强的溶解能力、与分子(或溶质)的相互作用小的溶剂。
本发明所述的“反溶剂”是指对分子(或溶质)的溶解能力低、差或不溶。
通过将良溶剂与反溶剂结合使用,从而降低待结晶物在溶剂中的溶解性。由于反溶剂与良结合,降低了分子(或溶质)的溶解性,从而使分子(或溶质)析出形成固相。然后从液相中过滤出固体,再分离两种溶剂,即可得到目标的结晶物。
本发明所述的“x-射线粉末衍射图谱或xrpd”是指根据布拉格公式2dsinθ=nλ(式中,λ为x射线的波长,
本发明所述的“差示扫描量热分析或dsc”是指在样品升温或恒温过程中,测量样品与参考物之间的温度差、热流差,以表征所有与热效应有关的物理变化和化学变化,得到样品的相变信息。
本发明所述的“2θ或2θ角度”是指衍射角,θ为布拉格角,单位为°或度,2θ的误差范围为±0.3或±0.2或±0.1。
本发明所述的“晶面间距或晶面间距(d值)”是指空间点阵选择3个不相平行的连结相邻两个点阵点的单位矢量a,b,c,它们将点阵划分成并置的平行六面体单位,称为晶面间距。空间点阵按照确定的平行六面体单位连线划分,获得一套直线网格,称为空间格子或晶格。点阵和晶格是分别用几何的点和线反映晶体结构的周期性,不同的晶面,其面间距(即相邻的两个平行晶面之间的距离)各不相同;单位为
本发明还涉及,包括式(i)所示的化合物的ii晶型,以及任选的一种或多种药用载体和/或稀释剂的药物组合物。所述药物组合物可以制成药学上可接受的任一剂型。例如,本发明的ii晶型或药物制剂可以配制为片剂、胶囊剂、丸剂、颗粒剂、溶液剂、混悬剂、糖浆剂、注射剂(包括注射液、注射用无菌粉末与注射用浓溶液)、栓剂、吸入剂或喷雾剂。
发明的有益效果
与现有技术相比,本发明制备的式(i)所示化合物的ii晶型纯度较高,在光照、高温、高湿的条件下晶型均未发生改变、晶型稳定性良好;hplc纯度变化小、化学稳定性高;本发明技术方案得到的式(i)所示化合物的ii晶型能够满足生产运输储存的药用要求,生产工艺稳定、可重复可控,能够适应于工业化生产。
附图说明
图1为式(i)所示化合物ii晶型的x-射线粉末衍射图谱。
图2为式(i)所示化合物ii晶型的dsc图谱。
图3为式(i)所示化合物c晶型的x-射线粉末衍射图谱。
图4为式(i)所示化合物c晶型的dsc图谱。
图5为ii晶型0天的xrpd图。
图6为ii晶型在40℃、相对湿度rh75%条件下放置15天后的xrpd图。
图7为c晶型0天的xrpd图。
图8为c晶型在40℃、相对湿度rh75%天条件下放置15天后的xrpd图。
具体实施方式
以下将结合实施例更详细地解释本发明,本发明的实施例仅用于说明本发明的技术方案,并非限定本发明的实质和范围。
试验所用仪器的测试条件:
1、差示扫描量热仪(differentialscanningcalorimeter,dsc)
仪器型号:mettlertoledodsc1staresystem
吹扫气:氮气
升温速率:10.0℃/min
温度范围:40-300℃
2、x-射线衍射谱(x-raypowderdiffraction,xrpd)
(1)仪器型号:brukerd8focusx-射线粉末衍射仪
射线:单色cu-kα射线(λ=1.5406)
扫描方式:θ/2θ,扫描范围:2-40°
电压:40kv,电流:40ma
(2)仪器型号:rigakuultimalvx-射线粉末衍射仪
射线:单色cu-kα射线(λ=1.5418)
扫描方式:θ/2θ,扫描范围:3-45°
电压:40kv,电流:40ma
3、动态水吸附仪(dynamicvapoursorption,dvs)
仪器型号:dvsadvantage
温度:25℃
溶剂:水
湿度变化:0-95-0-95-0%rh,dm/dt=0.002
实施例1
将300mg(s)-2-(4-(4-(4-(6-氟-5h-咪唑并[5,1-a]异吲哚-5-基)哌啶-1-基)苯基)-1h-吡唑-1-基)乙醇(按专利申请wo2016169421中的实施例40、41中的方法制备)粗品加入到反应瓶中,再加入二氯甲烷35ml,加热至回流,固体溶解,关闭加热,搅拌析晶。次日,抽滤,干燥得白色固体238mg,收率为79.3%。该结晶样品的x-射线衍射图谱见图1,其dsc谱图见图2,将此晶型定义为ii晶型,其特征峰位置如下表所示:
表1、ii晶型特征峰
实施例2
将300mg(s)-2-(4-(4-(4-(6-氟-5h-咪唑并[5,1-a]异吲哚-5-基)哌啶-1-基)苯基)-1h-吡唑-1-基)乙醇粗品加入到反应瓶中,加入四氢呋喃15ml,加热至回流,固体溶解,关闭加热,搅拌析晶。次日,抽滤,干燥得白色固体264mg,收率为88.0%。该结晶样品经xrpd检测确定产物为ii晶型。
实施例3
将300mg(s)-2-(4-(4-(4-(6-氟-5h-咪唑并[5,1-a]异吲哚-5-基)哌啶-1-基)苯基)-1h-吡唑-1-基)乙醇粗品加入到反应瓶中,加入乙二醇二甲醚45ml,加热至回流,固体溶解,关闭加热,搅拌析晶。次日,抽滤,干燥得白色固体185mg,收率为61.7%。该结晶样品经xrpd检测确定产物为ii晶型。
实施例4
将300mg(s)-2-(4-(4-(4-(6-氟-5h-咪唑并[5,1-a]异吲哚-5-基)哌啶-1-基)苯基)-1h-吡唑-1-基)乙醇粗品加入到反应瓶中,加入无水乙醇30ml,加热至回流,固体溶解,关闭加热,搅拌析晶。次日,抽滤,干燥得白色固体262mg,收率为87.3%。该结晶样品经xrpd检测确定产物为ii晶型。
实施例5
根据wo2016169421中实施例40、41的方法制备(s)-2-(4-(4-(4-(6-氟-5h-咪唑并[5,1-a]异吲哚-5-基)哌啶-1-基)苯基)-1h-吡唑-1-基)乙醇(即化合物41),经xrpd测定为晶型,该晶型在4.1,6.0,6.3,6.5,7.6,8.4,8.7),9.0,10.1,10.7,12.1,12.5,14.2,15.2,16.3,17.9,18.4,18.8,19.4,19.9,20.5,21.3,22.1,22.6,23.4,24.2,25.6,26.4,27.0,27.3,28.3,28.8,30.0和31.6处有特征峰,该晶型为c晶型。
实施例6
将实施例1所得的ii晶型与实施例5的c晶型敞口平摊放置,考察40℃、相对湿度rh75%条件下的稳定性。考察取样时间为15天,xrpd检测结果如说明书附图图5~图8所示。
试验结论:
由说明书附图图5~图8的稳定性考察结果表明式(i)所示化合物的ii晶型在40℃、相对湿度rh75%放置的条件下,xrpd峰型基本未发生变化,ii晶型稳定;c晶型在40℃、相对湿度rh75%放置的条件下,xrpd峰型发生了改变,部分峰型特征消失,结晶度降低;由此可见,在40℃、相对湿度rh75%放置的条件下ii晶型的物理稳定性优于c晶型。
- 还没有人留言评论。精彩留言会获得点赞!