一种环状RNA表达载体及使用所述表达载体的TRAP方法与流程
本发明属于生物技术领域,具体涉及一种环状rna表达载体及使用所述表达载体的适用于circrna互作rna或蛋白的trap方法。
背景技术
环状rna(circularrnas)是广泛且多样地存在于多种生物细胞中,具有调控基因表达作用的一类内源性ncrna分子。最早于20世纪70年代在rna病毒中被发现。之后几十年,人们在一些转录本中也发现了一些由外显子构成的circrna,如结肠直肠癌缺失基因转录本、人类ets-1基因转录本等,但当时它们被认为是转录的“噪音”,未引起人们太多的关注。而近年来,随着生物信息学技术的快速发展,salzman等、jeck等、memczak等及guo等分别在人类和小鼠的多种细胞中发现了成千上万种circrna。
近年来,随着高通量测序技术及基因芯片技术的高速发展,研究者们在真核生物体内发现了越来越多的circrna。如salzman等发现了数以百计的与人类基因表达相关的circrna。jeck等在人类成纤维细胞中检测到了高达25000多种circrna。
目前发现的circrna,根据其在基因组中的来源及其构成序列的不同,可以分为3类:外显子来源的环形rna分子,内含子来源的环形rna分子和由外显子和内含子共同组成的环形rna分子。
circrna具有以下特征:表达水平差异较大;具有序列保守性;偏好于细胞浆中;不易被核酸外切酶rnaser降解;外显子来源为主;属于ncrna;可充当cerna的角色调控靶基因的表达;在转录或转录后水平发挥调控作用。这些特征使得circrna在研究疾病发生机制、干预靶点和生物学标志物等方面显示显著优势。
研究表明,circrna在转录及转录后水平具有非常重要的基因表达调控作用。1、mirna海绵作用:小脑变性相关蛋白1反义转录物(antisensetothecerebellardegeneration-relatedprotin1transcript,cdr1as)是一种circrna,拥有至少60个与微小rna-7(microrna-7,mir-7)相结合的保守结合位点,从而充当mir-7海绵,有效影响mir-7靶基因活性。此外,研究发现,睾丸特异性基因sry的circrna具有16个微小rna-138的靶位点,可与mir-138相互作用,充当mir-138海绵。2、调控基因转录:zhang等鉴定出一种来源于基因内含子区域的circrna,发现这种circrna分子在细胞核内含量丰富,可参与基因转录调控。3、调控rna结合蛋白:bohjanen等设计了一种能够特异地结合反式激活调控蛋白(transactivatingregulatoryprotein,tat)的circrna分子,从而抑制i型人类免疫缺陷病毒1(hiv-1)基因的表达。4、参与部分蛋白质翻译:丁型肝炎病毒(hepatitisdvirusantigen,hdag)在疾病发展中起到重要的作用。另外,研究发现,人头骨瘤细胞u2os中,circrna具有翻译功能,尽管其翻译效率非常低。
近年来,随着高通量测序技术及基因芯片技术的高速发展,研究者们在真核生物体内发现了越来越多的circrna。如salzman等发现了数以百计的与人类基因表达相关的circrna。jeck等在人类成纤维细胞中检测到了高达25000多种circrna。danan等又发现了生物体内circrna的各种特性。同时,人们在认识circrna的结构时候,发现它们在疾病的发生和发展中发挥了重要作用。如circrna与肿瘤、神经系统疾病、动脉粥样硬化、糖尿病及病毒性肝炎等有关,因此,circrna作为分子标记物的诊断价值在疾病的发现和治疗中发挥着巨大作用。然而,circrna在生物体内发挥的功能及分子作用机制的研究却非常少。目前的研究中,仅仅明确了circrna具有吸附mirna的海绵作用、调控基因转录等作用。目前常用于研究circrna与蛋白或rna相互作用的方法rna反义纯化(rnaantisensepurification,rap)。
rna反义纯化(rnaantisensepurification,rap)是一种用于研究转录后水平鉴定rna与rna、rna与蛋白质互作机理的方法。rap技术首先用紫外交联固定细胞,以维持如circrna等调控rna与rna、rbp的相互作用,然后进行细胞裂解,接着用生物素标记的寡核苷酸探针(针对circrna的接头序列)与靶circrna杂交,基于生物素和链霉亲和素相互作用的原理,用链霉亲和素磁珠来分离、纯化探针-基因或蛋白质复合体,最后从纯化的复合体中分离蛋白质或rna,以进行下游的分析。
近年来,trap(taggedrnaaffinitypurification)技术也应用于研究rna互作上,即使用特定的rna标签分离表达的rna的方法。最广泛使用的标签之一ms2,是存在于ms2复制酶mrna的核糖体结合位点的19核苷酸长的病毒(噬菌体)rna序列,其折叠成发夹环结构。通过ms2噬菌体衣壳rna结合蛋白ms2(kd为3-300×109,取决于茎环序列和ms2rbp变体),以高特异性和亲和力识别该发夹环。作为含有肽标签的嵌合蛋白质的rbpms2的表达促进了[ms2rna/ms2蛋白]复合物与复合物中存在的其它分子的分离。ms2已被广泛用于标记在体外和体内转录其他应用的rna。
ms2下拉方法由三个基本步骤组成:
①涉及构建两个质粒载体并将其共转染入哺乳动物细胞。第一个质粒表达含有感兴趣的测试rna的嵌合rna,然后是几个ms2rna发夹(通常为12或24个串联ms2发夹环)。通常建议将ms2序列附着在测试rna的3'末端,但在poly(a)尾部之前,以避免阻断翻译或可能翻译ms2序列。对照质粒简单地表达没有感兴趣的测试rna的ms2发夹。第二质粒表达包含ms2衣壳蛋白和亲和标签的嵌合蛋白。与检测嵌合蛋白质相比,为了减少非特异性结合,表达更多的嵌合rna是重要的。例如表达用ms2发夹环(lincrna-p21-ms2)标记的小鼠lincrna-p21作为感兴趣的rna(ms2rna在对照平行转染中表达)。
②检测蛋白质ms2-gst涉及收获在转染质粒后24-48小时培养培养物,然后用保留天然rnp的完整性的缓冲液进行细胞裂解。然后将细胞裂解物与亲和试剂混合并使其结合,分离并洗涤rnp复合物。亲和试剂是附着于珠粒的谷胱甘肽-sh(gsh)gsh珠以高亲和力结合融合蛋白(ms2-gst)的gst组分,并可通过离心有效分离ms2rnp。然后用dna酶处理回收的沉淀物,分离rna进行进一步分析。
③鉴定纯化的rnp复合物中存在的rna或protein。
上述rap方法具有3点较明显的缺点:(1)实验中使用到探针与circrna杂交拉下circrna,杂交温度较高,杂交效率较低且rna易降解,容易造成实验结果的假阴性;(2)circrna接头序列可能与蛋白结合,造成探针无法与circrna结合,无法拉下circrna,造成实验无法进行;(3)需要合成带有标记的rna探针且探针易于降解。
而传统的trap实验目前只应用lncrna等互作rna或蛋白的研究,尚未应用到circrna的研究中,导致circrna的研究有很大的局限性。
技术实现要素:
为了解决现有技术存在的问题,本发明提供了一种环状rna表达载体及使用所述表达载体的适用于circrna互作rna或蛋白的trap方法,解决了传统的trap实验目前只应用lncrna等互作rna或蛋白研究的局限性问题。
本发明的目的是提供一种环状rna表达载体。
本发明的再一目的是提供一种使用所述表达载体的trap方法trap方法。
根据本发明的环状rna表达载体,包含启动子-反向互补反应上游元件-内含子ag受体(ms2部分序列)-多克隆位点-内含子gt供体(ms2部分序列)-反向互补反应下游元件-终止子,所述内含子ag受体的序列如seqidno:1所示,所述内含子gt供体的序列如seqidno:2所示。
所述seqidno:1的序列为:gatcacccatgtctgcag。
所述seqidno:2的序列为:ctctagaaaacatgaggtaag。
根据本发明的环状rna表达载体,其中,所述反向互补反应上游元件的序列如seqidno:3所示,所述反向互补反应下游元件的序列如seqidno:4所示。
所述seqidno:3的序列为:
ggtggactgcctgaggtcaggagttcgtgaccagactgaccaacatggtgaaaccctgtctctactaaaaatacaaaaaaaattagccaggtgcggtggcatgcacctgtaatcccagctactcgggaggctaaggcaggggaattgcttgaaccagggaggtggaggtcgcagtgagccgagatggcgccactgcactccagcctgggcaacagagagagactctatcttaaaaaaaaaaaaaaaaattattctggtaggctcaggccccatgtggcct。
所述seqidno:4的序列为:
aggccacatggggcctgagcctaccagaataatttttttttttttttttaagatagagtctctctctgttgcccaggctggagtgcagtggcgccatctcggctcactgcgacctccacctccctggttcaagcaattcccctgccttagcctcccgagtagctgggattacaggtgcatgccaccgcacctggctaattttttttgtatttttagtagagacagggtttcaccatgttggtcagtctggtcacgaactcctgacctcaggcagtccaccgctcgtggcttttttttttttttttttttttttttgagacagagtatcaccctgtcacccagg。
根据本发明的环状rna表达载体,其中,所述启动子为pcmv启动子;所述终止子为bghpa终止子。
根据本发明的环状rna表达载体,其中,所述多克隆位点依此包含bamhi、填充序列和ecori。
根据本发明的trap方法,所述trap方法使用所述环状rna表达载体。
根据本发明的trap方法,其中,所述的trap方法包含以下步骤:
a、环状rna表达载体的构建:得到pms2-环状rna表达载体;
b、细胞培养、转染和裂解:准备细胞,使用步骤a得到的所述pms2-环状rna表达载体或pms2空载转染细胞,培养后进行裂解,得到裂解液;
c、取步骤b中的裂解液,进行pulldown实验,得到nt2缓冲液,标记为trap液;
d、取步骤c中的trap液,依此进行rnp收集和rna纯化,并进行逆转录和qpcr检测。
根据本发明的trap方法,其中,步骤b、细胞培养、转染和裂解的具体操作为:
①准备细胞;
②共同转染步骤a得到的所述pms2-环状rna表达载体或pms2空载,和用150μloptimem稀释的1μgpms2gst;
③48小时后,用pbs洗涤细胞两次,并在lysisbuffer,蛋白酶抑制剂,rnase抑制剂和100mmdtt裂解液中裂解在冰上10分钟;
④通过刮擦收集细胞裂解物后,在4℃下以10,000×g离心裂解物15分钟;
⑤使用bradford测定法收集上清液并测量蛋白质浓度;
⑥使用2μg/μl裂解液进行pulldown实验。
根据本发明的trap方法,其中,步骤c、pulldown实验的具体操作为:
①分别取200μl10%的gsh磁珠,用冰冷的pbs洗涤gsh磁珠两次,并用等体积的pbs重悬,制成50%浆液;
②将上述磁珠分别加入两组上清液,4℃孵育3小时;
③4℃,4,500rpm离心1分钟后,用1mlnt2缓冲液洗涤珠子两次;
④分别将磁珠与20u无rna酶的dna酶i在37℃下在100μl反应体积中孵育15分钟;
⑤加入1mlnt2缓冲液;
⑥若后续实验同时研究蛋白和rna,则1mlnt2缓冲液平均分为两管,4℃,4500rpm离心1分钟;
⑦若后续实验只研究蛋白,则1mlnt2缓冲液按100μl及900μl分为两管,分别标记为traprna,trapprotein,4℃,4500rpm离心1分钟;
⑧若后续实验只研究rna,则1mlnt2缓冲液按100μl及900μl分为两管,分别标记为trapprotein,traprna,4℃,4500rpm离心1分钟。
根据本发明的trap方法,其中,步骤d中rnp收集的具体操作为:
①分别将trapprotein组珠子加入30μlproteinelutionbuffer1,7μlproteinelutionbuffer2及0.3μldtt,置于沸水中煮沸10min,中间间隔混匀,磁力架上移上清至新离心管中;
②重复步骤①一次;
③蛋白质样品置于-80℃保存备用;
④取15μl蛋白样品开展page-银染检测;
⑤取15μl蛋白样品开展page-westernblot检测;
⑥取30μl蛋白样品开展lc-ms检测;
⑦剩余样品-20℃保存备用。
根据本发明的trap方法,其中,步骤d中rna纯化的具体操作为:
①分别将traprna组珠子加入200μlrnaelutionbuffer,2μl蛋白酶k,inputrna各加入100μlrnaelutionbuffer,2μl蛋白酶k,在55℃下孵育30分钟;
②4℃,10,000g离心5分钟收集上清液;
③分别加入等洗脱体积的苯酚-氯仿-异戊醇混合液到inputrna,traprna中,苯酚-氯仿-异戊醇的体积比为25:24:1;涡旋混匀5min;
④13,000g4℃离心10分钟,收集上层水相,转移到新无rna酶离心管中;
⑤200μl上清液分别加入400μl100%乙醇,20μl乙酸钠和2μlglycogen混合;
⑥在-20℃放置2小时后,在4℃下以10,000×g离心20分钟;
⑦用500μl70%乙醇洗涤得到的rna沉淀,10,000×g,4℃离心10分钟;
⑧干燥rna颗粒,并重悬于10-20μl无rnase的水中;
⑨进行逆转录和qpcr检测。
本发明的有益效果为:
1、本发明提供了一种环状rna表达载体,将ms2rna分成两部分,分别位于circrna成环前的两端,只有circrna成环时才能形成可被ms2蛋白识别的ms2rna,避免了线性rna对实验的干扰。
2、所述trap方法使用所述环状rna表达载体,本发明将传统的trap实验进行改进并应用circrna的研究上,为circrna的研究提供了新的方法。
3、所述trap方法避免使用了探针杂交的方法,彻底不受接头序列被蛋白结合后探针不能结合circrna对circrna研究存在假阴性的影响,且降低了探针杂交过程rna的降解。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
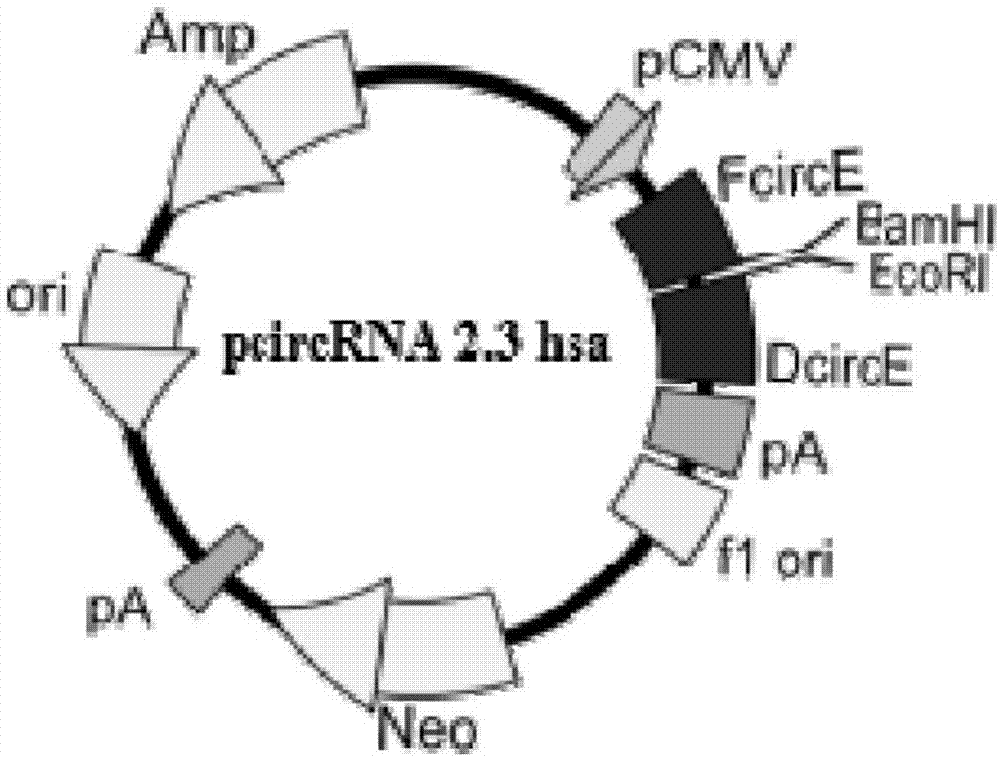
图1是pcircrna2.3hsa表达载体模式图;
图2是pcircrna2.2hsa表达载体模式图;
图3是所述trap方法和rap方法的富集效果比对图;
图4是所述trap方法和rap方法对circ-foxo3靶蛋白cdk2富集效率比对图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将对本发明的技术方案进行详细的描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施方式,都属于本发明所保护的范围。
实施例1
本发明构建了含pcmv启动子-反向互补反应上游元件(reversecomplementarymatches-upstream,rcms-up)-ms2部分序列-多克隆位点-ms2部分序列-反向互补反应下游元件(reversecomplementarymatches-downstream,rcms-down)-bghpa为主体架构的circrna形成真核表达载体,并经过测序验证。该质粒载体及慢病毒载体分别命名为pcircrna2.3hsa、pcircrna2.2hsa。
pcircrna2.3hsa、pcircrna2.2hsa的表达载体模式图如图1和图2所示。
所述环状rna表达载体核心元件序列如下:
所述反向互补反应上游元件的序列如seqidno:3所示;
所述内含子ag受体(ms2部分序列)的序列如seqidno:1所示;
所述多克隆位点的序列如seqidno:5所示;
seqidno:5的序列为:ggatccgcgcaggaccggaattc;
所述内含子gt供体(ms2部分序列)的序列如seqidno:2所示;
所述反向互补反应下游元件的序列如seqidno:4所示。
所述环状rna表达载体,将ms2rna分成两部分,分别位于circrna成环前的两端,只有circrna成环时才能形成可被ms2蛋白识别的ms2rna,避免了线性rna对实验的干扰。
环状rna表达载体,是以hsa_circ_0001946为研究目标得到的,hsa_circ_0001946为外显子circrna,其序列如seqidno:6所示。
实施例2
一种trap方法,方法包含以下步骤:
a、环状rna表达载体的构建:按照实施例1中的序列,采用基因合成的方式合成得到环状rna表达载体;
b、细胞培养,转染和裂解物的制备:
①准备2×107细胞;
②共同转染2μg质粒pms2-circrna-0001946或pms2,和用150μloptimem稀释的1μgpms2gst;
③48小时后,用pbs洗涤细胞两次,并在1.2mllysisbuffer,12μl蛋白酶抑制剂,12μlrnase抑制剂和12μl100mmdtt裂解液中裂解在冰上10分钟;
④通过刮擦收集细胞裂解物后,在4℃下以10,000×g离心裂解物15分钟;
⑤使用bradford测定法收集上清液并测量蛋白质浓度;
⑥使用1000μl裂解液(2μg/μl)进行pulldown实验;
c、pulldown实验:
①分别取200μl10%的gsh磁珠,用冰冷的pbs洗涤gsh磁珠两次,并用等体积的pbs重悬,制成50%浆液;
②将上述磁珠分别加入两组上清液,4℃孵育3小时;
③4℃,4,500rpm离心1分钟后,用1mlnt2缓冲液洗涤珠子两次;
④分别将磁珠与20u无rna酶的dna酶i在37℃下在100μl反应体积中孵育15分钟;
⑤加入1mlnt2缓冲液;
⑥若后续实验同时研究蛋白和rna,则1mlnt2缓冲液平均分为两管,4℃,4500rpm离心1分钟;
⑦若后续实验只研究蛋白,则1mlnt2缓冲液按100μl及900μl分为两管,分别标记为traprna,trapprotein,4℃,4500rpm离心1分钟;
⑧若后续实验只研究rna,则1mlnt2缓冲液按100μl及900μl分为两管,分别标记为trapprotein,traprna,4℃,4500rpm离心1分钟;
d、rnp收集:
①分别将trapprotein组珠子加入30μlproteinelutionbuffer1,7μlproteinelutionbuffer2及0.3μldtt,置于沸水中煮沸10min,中间间隔混匀,磁力架上移上清至新离心管中;
②重复步骤①一次;
③蛋白质样品置于-80℃保存备用;
④取15μl蛋白样品开展page-银染检测;
⑤取15μl蛋白样品开展page-westernblot检测;
⑥取30μl蛋白样品开展lc-ms检测;
⑦剩余样品-20℃保存备用;
e、rna纯化:
①分别将traprna组珠子加入200μlrnaelutionbuffer,2μl蛋白酶k,inputrna各加入100μlrnaelutionbuffer,2μl蛋白酶k,在55℃下孵育30分钟;
②4℃,10,000g离心5分钟收集上清液;
③分别加入等洗脱体积的苯酚-氯仿-异戊醇混合液到inputrna,traprna中,苯酚-氯仿-异戊醇的体积比为25:24:1;涡旋混匀5min;
④13,000g4℃离心10分钟,收集上层水相,转移到新无rna酶离心管中;
⑤200μl上清液分别加入400μl100%乙醇,20μl乙酸钠和2μlglycogen混合;
⑥在-20℃放置2小时后,在4℃下以10,000×g离心20分钟;
⑦用500μl70%乙醇洗涤得到的rna沉淀,10,000×g,4℃离心10分钟;
⑧干燥rna颗粒,并重悬于10-20μl无rnase的水中;
⑨进行逆转录和qpcr检测。
实施例3
本实施例为与现有技术相比,本发明提供的trap方法的对比试验。
1、通过不同技术对circrna的富集,将hek293细胞裂解蛋白,分别使用本发明所述的trap方法和现有的rap方法对hsa_circ_0001946进行富集,结果如图3所示。
需要说明的是,转化为黑白灰度图之后,图3的个别颜色之间区别较小,不容易分辨,在此做出解释说明:横坐标的每个系列,均按照hsa_circ_0001946、cdr1、β-actin、mir-7、u6的顺序排列。
图3对应的实验参数为:hsa_circ_0001946:作为trap实验组;ms2:trap实验阴性对照组;hsa_circ_0001946:rap实验组;lacz:rap实验阴性对照组。
从图3中能够看出,本发明所述的trap方法对于hsa_circ_0001946的富集效率明显高于rap方法,同时检测hsa_circ_0001946宿主基因的富集效率,本发明所述的trap方法中只有成环的rna才能形成ms2rna,并且被gst-ms2识别,并通过gsh磁珠拉下来,所以基本不含有宿主基因的残留,而现有的rap方法是基于探针杂交的过程,存在着一定的宿主基因的残留干扰实验;并且,trap方法中hsa_circ_0001946靶基因的富集效率也相应提高,rap是基于探针与rna杂交,若探针结合的位置有蛋白结合,会大大较低探针的效率,导致的假阴性结果,而trap方法是基于ms2rna与ms2蛋白结合,避免了这种假阴性结果。
综上,本试验说明本发明所述的trap方法与现有的rap方法相比,富集效率更高,非特异型结合更低,结果更准确。
2、通过不同技术对circrna的富集,将hek293细胞裂解蛋白,分别使用本发明所述的trap方法和现有的rap方法对circ-foxo3靶蛋白cdk2进行富集,结果如图4所示。
图4对应的实验参数为:input:10%input蛋白;circ-foxo3:实验组;nc:阴性对照组。
图4中a、b为本发明所述的trap方法的效果图;c为按照rap方法效果图,从图4中能够看出,trap方法相对于rap方法条带更深,蛋白产率更高,表明trap方法与现有的rap方法相比,具有更高的蛋白富集效率,特异性更好,蛋白富集效率更高,灵敏度高。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!