一种三蝶烯类离子液体功能材料、其制备及其应用的制作方法
本发明涉及一种三蝶烯类离子液体功能材料、其制备及其应用,属于气相色谱技术领域。
背景技术:
目前气相色谱(gc)分析中,商品柱种类有限,并且对于性质相近的物质(如难分离的异构体混合物)的分离能力存在一定限制,不能满足复杂样品色谱分析测定的要求。因而,设计合成新型高选择性gc固定相,用于高效分离难分离组分及其色谱分析测定,满足目前各行业日益增长的色谱分析测定需求具有重要应用价值。
蝶烯类材料具有良好的热稳定性和溶解性,易于功能化,是一类具有独特三维刚性结构和富电子空腔的桥环化合物,近年来在材料化学和气体分离等领域有广泛的应用。三蝶烯衍生物在聚合物气体膜分离、样品前处理和色谱固定相等领域已有报道。其中,液相色谱领域所涉及的是三蝶烯衍生物键合硅胶填充柱用于手性分离;而气相色谱领域所涉及的三蝶烯衍生物为弱极性固定相,对弱极性分子有着特殊选择性,但对极性分子和难分异构体的分离、选择有着很大的局限。因此,需要对三蝶烯进一步衍生使其对极性分子和难分异构体也有很好的分离效果。
技术实现要素:
针对现有gc固定相对于性质相近的物质的分离能力差的问题,本发明提供了一种三蝶烯类离子液体功能材料,以三蝶烯结构单元为母体,在三蝶烯的苯环上通过化学键合连接有机阳离子结构单元,该功能材料用于gc固定相对于性质相近组分表现出高选择性,能够高效分离出不同极性各类组分混合物,满足色谱分析测定需求。
本发明的目的是通过以下技术方案实现的。
一种三蝶烯类离子液体功能材料,所述功能材料包含以下三种结构:
(1)
其中,r2+为含有氮或磷元素的阳离子基团,优选咪唑阳离子、苯并咪唑阳离子、胍盐阳离子、鏻盐阳离子、吡啶阳离子或铵盐阳离子;r1为o或c=o;x-为无机阴离子或有机阴离子,优选卤素离子、ntf2-、tfo-或pf6-;n=1~8,m=2~12。
(2)
其中,两个取代基团结构相同,r2+为含有氮或磷元素的阳离子基团,优选咪唑阳离子、苯并咪唑阳离子、胍盐阳离子、鏻盐阳离子、吡啶阳离子或铵盐阳离子;r1为o;x-为无机阴离子或有机阴离子,优选卤素离子、ntf2-、tfo-或pf6-;n=1~8,m=2~12。
(3)
其中,三个取代基团相同,r2+为含有氮或磷元素的阳离子基团,优选咪唑阳离子、苯并咪唑阳离子、胍盐阳离子、鏻盐阳离子、吡啶阳离子或铵盐阳离子;r1为o或c=o;x-均为无机阴离子或有机阴离子,优选卤素离子、ntf2-、tfo-或pf6-;n=1~8,m=2~12。
第(1)种结构的三蝶烯类离子液体功能材料的制备分为两种情况:
(a)r1为c=o时
1)按照不低于1:1的摩尔比将三蝶烯和无水氯化铝加入到有机溶剂中,再在-15℃~-10℃下滴加1.1~1.5倍于三蝶烯摩尔量的乙酰氯,反应不低于40min,分离提纯,得2-乙酰基三蝶烯。
2)按照不低于1:1的摩尔比将2-乙酰基三蝶烯与卤化铜加入到有机溶剂中,加热回流至不再产生气体和沉淀,分离提纯,得到卤代2-乙酰基三蝶烯。
3)按照不低于1:1的摩尔比将卤代2-乙酰基三蝶烯与r2cm+1h2m+3加入到有机溶剂中,回流反应不低于24h,冷却,洗涤,得到[tp-r2]x,即x-为卤素离子的三蝶烯类离子液体功能材料;其中,r2cm+1h2m+3优选为丁基咪唑、戊基咪唑、十二烷基苯并咪唑或1,1,3,3-四甲基-2-丁基胍。
4)[tp-r2]x与阴离子交换剂在有机溶剂中搅拌反应不低于24h,洗涤,得到tp-il,即x-不为卤素离子的三蝶烯类离子液体功能材料;其中,[tp-r2]x与阴离子交换剂摩尔比不低于1:1.1。
(b)r1为o时
1)先将三蝶烯加入到有机酸溶剂中,再滴加市售浓硝酸,在60℃~80℃下搅拌反应4h~8h,冷却析出后,分析提纯,得到2-硝基三蝶烯;其中,有机酸优选冰醋酸,12ml市售浓硝酸对应1g三蝶烯,优选采用洗脱剂在硅胶柱上进行分离提纯,洗脱剂优选体积比为4:1的石油醚和二氯甲烷的混合溶液。
2)在氮气保护以及雷尼镍催化剂作用下,2-硝基三蝶烯和水合肼在有机溶剂中于50℃~65℃下反应2h~4h,淬灭水合肼后,冷却,分离提纯,得到产物2-氨基三蝶烯;其中,有机溶剂优选四氢呋喃,优选1ml水合肼的量对应1mmol2-硝基三蝶烯,优选采用洗脱剂在硅胶柱上进行分离提纯,洗脱剂优选体积比为1:20的石油醚和甲醇的混合溶液。
3)将2-氨基三蝶烯溶于质量分数为10%~15%的硫酸溶液中,充分搅拌使溶液由浑浊变为澄清后,再加入1.1倍于2-氨基三蝶烯摩尔量的亚硝酸钠溶液,搅拌均匀后转移至回流的质量分数为50%~70%的硫酸溶液中,继续回流不少于2h,分离提纯,得到2-羟基三蝶烯;其中,优选采用洗脱剂在硅胶柱上进行分离提纯,洗脱剂优选体积比为4:1的石油醚和乙酸乙酯混合溶液。
4)在氮气保护下,2-羟基三蝶烯、cnh2nx2和碳酸钾按照1:x:y的摩尔比于有机溶剂中回流反应不低于40h,分离提纯,得tp-x;其中,x≥1.1,y≥1.5,有机溶剂优选为乙腈,cnh2nx2(二卤代烷)优选为1,4-二溴丁烷或1,8-二溴辛烷,优选采用洗脱剂在硅胶柱上进行分离提纯,洗脱剂优选体积比为1:8的为石油醚和乙酸乙酯混合溶液。
5)tp-x与r2cm+1h2m+3在有机溶剂中回流反应不少于24h,再用洗液洗涤,得到[tp-r2]x,即是x-为卤素离子的三蝶烯类离子液体功能材料;其中,tp-x与r2cm+1h2m+3的摩尔比不低于1:1.1,r2cm+1h2m+3优选为丁基咪唑、戊基咪唑、十二烷基苯并咪唑或1,1,3,3-四甲基-2-丁基胍,有机溶剂优选为乙腈,洗液优选为乙醚。
6)tp-r2与阴离子交换剂在溶剂中搅拌反应不低于24h,制得tp-il,即x-不为卤素离子的三蝶烯类离子液体功能材料;其中,tp-r2与阴离子交换剂的摩尔比不低于1:1.1,有机溶剂优选为二氯甲烷。
第(2)种结构的三蝶烯类离子液体功能材料的制备步骤如下:
1)蒽与对苯醌按摩尔比1:1~1:1.5溶解于有机溶剂中,加热回流不少于3h,冷却,分离纯化,制得三蝶烯对苯二醌;其中,选用沸点130℃~140℃的有机溶剂,如二甲苯。
2)将三蝶烯对苯二醌溶解于冰醋酸溶剂中,加热回流不少于1h,再滴加与三蝶烯对苯二醌等摩尔量的hbr,继续反应不少于0.5h,冷却后收集固体产物,制得三蝶烯对苯二氢醌(tp-2oh)。
3)在n2保护下,三蝶烯对苯二氢醌、cnh2nx2和碳酸钾按照1:x:y的摩尔比于有机溶剂中回流反应不低于30h,冷却,分离纯化,得到tp-2x;其中,x≥3,y≥2,cnh2nx2(二卤代烷)优选为1,4-二溴丁烷或1,8-二溴辛烷。
4)tp-2x与r2cm+1h2m+3按摩尔比不低于1:2溶解于有机溶剂中,加热回流不少于24h,分离纯化,得到[tp-2r2]x2,即是x-为卤素离子的三蝶烯类离子液体功能材料;其中,r2cm+1h2m+3为含烷基链的氮或磷基团,优选为丁基咪唑、戊基咪唑、十二烷基苯并咪唑或1,1,3,3-四甲基-2-丁基胍。
5)[tp-2r2]x2与阴离子交换剂在有机溶剂中搅拌反应不少于24h,洗涤,制得tp-2il,即x-不为卤素离子的三蝶烯类离子液体功能材料;其中,tp-2r2]x2与阴离子交换剂的摩尔比不小于1:2.2。
第(3)种结构的三蝶烯类离子液体功能材料的制备分为如下两种情况:
(a)r1为o时
1)三蝶烯和市售浓硝酸于60℃~90℃下反应20h~40h,冷却析出,粗产物分离提纯,得到2,6,14-三硝基三蝶烯;
2)在氮气保护以及催化剂作用下,2,6,14-三硝基三蝶烯和产氢还原剂于含有无水甲醇的有机溶剂中反应12h~24h,分离提纯,得到2,6,14-三氨基三蝶烯;其中,2,6,14-三硝基三蝶烯与产氢还原剂的摩尔比不小于1:5,产氢还原剂优选硼氢化钾或硼氢化钠,催化剂为金属钯催化剂,无水甲醇的摩尔量大于产氢还原剂的摩尔量少于溶剂的摩尔量,优选10ml无水甲醇对应1g2,6,14-三硝基三蝶烯。
3)将2,6,14-三氨基三蝶烯溶于质量分数为10%~15%的硫酸溶液中,充分搅拌使溶液由浑浊变为澄清后,再加入亚硝酸钠溶液,搅拌均匀后转移至回流的质量分数为50%~70%的硫酸溶液中,继续回流不少于2h,分离提纯,得到2,6,14-三羟基三蝶烯;其中,亚硝酸钠与2,6,14-三氨基三蝶烯的摩尔比为3.3:1。
4)在氮气保护下,2,6,14-三羟基三蝶烯、cnh2nx2和碳酸钾按照1:x:y的摩尔比于有机溶剂中回流反应不低于40h,分离提纯,得三蝶烯三取代衍生物;其中,x≥4,y≥4,cnh2nx2(二卤代烷)优选为1,4-二溴丁烷或1,8-二溴辛烷。
5)三蝶烯三取代衍生物与r2cm+1h2m+3在有机溶剂中回流反应不低于2天,制得[tp-3r2]x3,即x-为卤素离子的三蝶烯类离子液体功能材料;其中,三蝶烯三取代衍生物与r2cm+1h2m+3的摩尔比不低于1:3.3,r2cm+1h2m+3优选为丁基咪唑、戊基咪唑、十二烷基苯并咪唑或1,1,3,3-四甲基-2-丁基胍。
6)[tp-3r2]x3与阴离子交换剂在有机溶剂中搅拌反应不低于24h,得到tp-3il,即x-不为卤素离子的三蝶烯类离子液体功能材料;其中,[tp-3r2]x3与阴离子交换剂的摩尔比不小于1:3.3。
(b)r1为c=o时
1)将三蝶烯和无水氯化铝按1:4~1:6的摩尔比加入到有机溶剂中,再在-15℃~-10℃下滴加3.5~7倍于三蝶烯摩尔量的溴代烷酰氯,然后在10℃~30℃下搅拌反应不低于4h,分离提纯,得2,6,14-三卤代烷酰基三蝶烯;其中,有机溶剂优选二氯甲烷,溴代烷酰氯结构式为
2)2,6,14-三卤代烷酰基三蝶烯与r2cm+1h2m+3在有机溶剂中回流反应不少于24h,冷却,洗涤,得到[tp-3r2]x3,即x-为卤素离子的三蝶烯类离子液体功能材料;其中,2,6,14-三卤代烷酰基三蝶烯与r2cm+1h2m+3的摩尔比不小于1:3.3,r2cm+1h2m+3优选为丁基咪唑、戊基咪唑、十二烷基苯并咪唑或1,1,3,3-四甲基-2-丁基胍,有机溶剂优选乙腈,洗涤剂优选石油醚和乙醚的混合液。
3)[tp-3r2]x3与阴离子交换剂在有机溶剂中搅拌反应不低于24h,洗涤,得到tp-3il,即x-不为卤素离子的三蝶烯类离子液体功能材料;其中,[tp-3r2]x3与阴离子交换剂按摩尔比不低于1:3.3,有机溶剂优选二氯甲烷。
所述阴离子交换剂是含有x-的化合物,且此时x-不包含卤素离子。x-为ntf2-、tfo-或pf6-时,相应地,阴离子交换剂为双(三氟甲基磺酰)亚胺锂,三氟甲磺酸锂,六氟磷酸锂。
将本发明所述三蝶烯类离子液体功能材料涂渍到毛细管柱内壁表面后,可以用于气相色谱分析。
有益效果:
(1)本发明所述的三蝶烯类离子液体功能材料具有三蝶烯母体、阳离子功能化单元和阴离子结构单元的各自性能优势,作为气相色谱固定相对性质相近组分表现出高选择性,可以高效分离不同极性各类组分混合物,尤其是难分离的各类异构体混合物,相比市售色谱柱表现出明显的分离优势。
(2)本发明所述的三蝶烯类离子液体功能材料在有机溶剂中具有良好的溶解性和成膜性,易于制备高柱效的色谱柱;而且采用此类功能材料作为gc固定相,分离性能具有良好的重复性、重现性和热稳定性,能够用于色谱分析测定。
(3)本发明所述的三蝶烯类离子液体功能材料其原料成本低廉、实验装置简单易得、合成方法简单、产品产率高。
附图说明
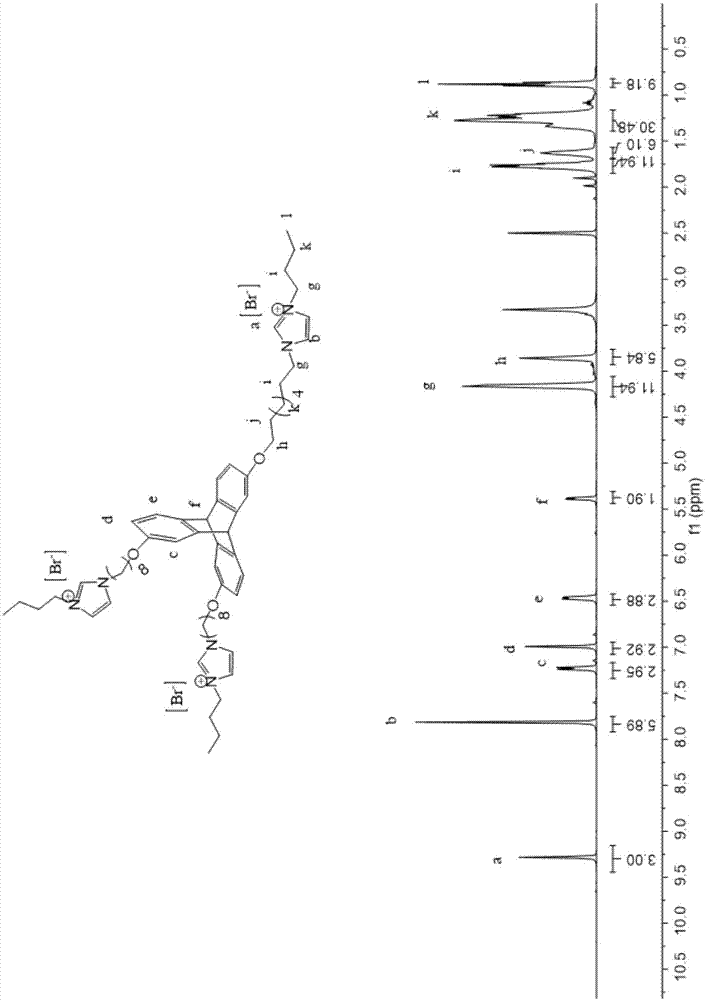
图1为实施例1中制备的[tp-2r2]x2的1h-nmr图谱。
图2为实施例1中制备的[tp-2r2]x2的13c-nmr图谱。
图3为实施例1中制备的tp-2il的1h-nmr图谱。
图4为实施例1中制备的tp-2il的13c-nmr图谱。
图5是基于实施例1中制备的tp-2il为固定相分离苯胺类异构体样品的色谱图;其中,1~10的色谱峰对应的物质依次为苯胺、邻甲苯胺、对甲苯胺、间甲苯胺、2,6-二甲苯胺、2,4-二甲苯胺、2,5二甲苯胺、3,5-二甲苯胺、2,3-二甲苯胺以及3,4-二甲苯胺。
图6是基于实施例2中制备的tp-2il固定相分离氯硝基苯异构体(a)和二氯苯异构体(b)的色谱图;其中,(a)图中1~3的色谱峰对应的物质依次为间氯硝基苯、对氯硝基苯以及邻氯硝基苯;(b)图中1~3的色谱峰对应的物质依次为对二氯苯、间二氯苯以及邻二氯苯。
图7是基于实施例3中制备的tp-3il为固定相分离不同类型组分样品的色谱图;其中,1~10的色谱峰对应的物质依次为癸烷、十一烷、苯乙醚、辛酮、苯甲醛、正辛醇、苯甲腈、苯胺、1,3-二溴苯、1,4-二甲氧基苯。
图8为实施例4中制备的[tp-3r2]x3的1h-nmr图谱。
图9为实施例4中制备的tp-3il的1h-nmr图谱。
图10是基于实施例4中制备的tp-3il为固定相分离不同类型组分样品的色谱图;其中,1~10的色谱峰对应的物质依次为丁基苯、正十二烷、壬酮、对二溴苯、癸酸甲酯、1,4-二甲氧基苯、萘、十一醇、对甲酚以及对氯苯酚。
图11是基于实施例5中制备的tp-il为固定相分离庚烷异构体(a)和苯二酚异构体(b)样品的色谱图;其中,(a)图中1~3的色谱峰对应的物质依次为2,2,3-三甲基丁烷、2,3-二甲基戊烷以及正庚烷;(b)图中1~5的色谱峰对应的物质依次为2,6-二甲苯酚、2,5-二甲苯酚、2,3-二甲苯酚、3,5-二甲苯酚以及3,4-二甲苯酚。
具体实施方式
下面结合附图和具体实施方式对本发明作进一步阐述,其中,所述方法如无特别说明均为常规方法,所述原材料如无特别说明均能从公开商业途径而得。
采用实施例中所制备的三蝶烯类离子液体功能材料作为固定相,制备毛细管色谱柱的步骤如下:
取一定长度(5m~30m)内径为0.25mm的石英毛细管柱,先用二氯甲烷冲洗20min,再在氮气气氛下于260℃老化3h;然后,在氮气压力下,向毛细管柱内连续通入氯化钠甲醇的饱和溶液,直至流出的液体呈浑浊为止,并将毛细管柱内溶液排出,再在氮气下于200℃保持3h;将实施例中所制备的三蝶烯类离子液体功能材料溶于二氯甲烷中,配制成适宜浓度(0.10mg/ml~0.50mg/ml,w/v)的固定相溶液,超声处理5min后,将固定相溶液通入毛细管柱中,将毛细管柱一端密封,另一端连接真空泵,在40℃的水浴中使溶剂蒸发,固定相沉积在毛细管柱内壁上,随后将该毛细柱进行老化,老化条件为:先在40℃下保持30min,再以1℃/min升温速率升温到180℃并保持6h,老化后的毛细管柱即为可用于gc分析测定的色谱柱。
实施例1
三蝶烯二取代化合物tp-2il咪唑类离子液体具体制备步骤如下:
(1)蒽与对苯醌按照1:1.2的摩尔比溶解于二甲苯中,并在140℃下回流5h,然后在0℃下冷却12h,过滤得到固体,再将固体用沸水洗涤,并在二甲苯中重结晶,再依次用二甲苯、石油醚洗涤,制得三蝶烯对苯二醌。
(2)将三蝶烯对苯二醌溶解于冰醋酸中,加热回流1h后,缓慢滴加与三蝶烯对苯二醌等摩尔量的hbr,产生沉淀,继续反应0.5h,冰浴冷却,过滤并收集固体产物,即制得三蝶烯对苯二氢醌(tp-2oh)。
tp-2oh的核磁共振氢谱的表征结果:1hnmr(400mhz,丙酮)δ7.83(s,2h),7.46–7.37(m,4h),7.02–6.93(m,4h),6.39(s,2h),5.93(s,2h)。
(3)按照1:4:3的摩尔比将tp-2oh、1,8-二溴辛烷和碳酸钾加入乙腈中,在n2保护下回流48h后,冷却,过滤,二氯甲烷洗涤,并用洗脱剂(v石油醚:v二氯甲烷=2:1)在硅胶柱上分离纯化,得到白色固体产物tp-2br。
tp-2x的核磁共振氢谱的表征结果:1hnmr(400mhz,cdcl3)δ7.46–7.33(m,4h),7.06–6.90(m,4h),6.48(s,2h),5.86(s,2h),3.93(t,j=6.4hz,4h),3.42(q,j=6.9hz,4h),1.94–1.79(m,8h),1.48–1.32(m,16h)。
(4)按照1:2.2的摩尔比将tp-2br和丁基咪唑加入乙腈中,于80℃下回流24h,旋蒸除去溶剂,再溶于二氯甲烷,然后在乙醚中沉淀,超声多次洗涤固体,得到产物[tp-2r2]x2。
[tp-2r2]x2的核磁共振谱图的表征结果:1hnmr(400mhz,dmso,400mhz,298k)δ9.29(s,2h),7.83(d,j=1.6hz,4h),7.57–7.27(m,4h),7.12–6.89(m,4h),6.61(s,2h),5.82(s,2h),4.18(q,j=6.8hz,8h),3.94(t,j=6.2hz,4h),1.97–1.16(m,32h),0.89(t,j=7.3hz,6h),如图1所示;13cnmr(dmso,400mhz,298k)δ(ppm):147.9,145.7,136.3,135.2,125.2,124.1,122.9,111.5,69.4,49.3,31.8,29,28.8,26.1,25.9,19.2,13.7,如图2所示。
(5)按照1:2.2的摩尔比将[tp-2r2]x2与阴离子交换剂lintf2加入二氯甲烷中,室温下搅拌24h,用去离子水洗涤产物,直至洗涤液中滴加硝酸银不再产生沉淀,得到产物tp-2il。
tp-2il的核磁共振谱图的表征结果:1hnmr(400mhz,dmso)δ9.29(s,2h),7.83(d,j=1.6hz,4h),7.57–7.27(m,4h),7.12–6.89(m,4h),6.61(s,2h),5.82(s,2h),4.18(q,j=6.8hz,8h),3.94(t,j=6.2hz,4h),1.97–1.16(m,32h),0.89(t,j=7.3hz,6h),如图3所示;13cnmr(dmso,400mhz,298k)δ(ppm):147.9,145.7,136.3,135.2,125.2,124.1,122.9,121.5,119.2,111.5,69.4,49.3,31.8,29,28.8,26.1,25.9,19.2,13.7,如图4所示。
将所制备的tp-2il溶于二氯甲烷中,配制成浓度为2.5mg/ml的固定相溶液,制备石英毛细管色谱柱(10m)。采用该色谱柱对苯胺、邻甲苯胺、对甲苯胺、间甲苯胺、2,6-二甲苯胺、2,4-二甲苯胺、2,5二甲苯胺、3,5-二甲苯胺、2,3-二甲苯胺以及3,4-二甲苯胺组成的苯胺类异构体样品进行分离,分离结果详见图5;其中,色谱条件:70℃~160℃,5℃/min,载气(氮气)流速0.8ml/min。由图可见,tp-2il固定相不仅高效分离了各个异构体组分,而且得到的色谱峰峰形对称性良好,表现出很好的色谱分离性能。此外,tp-2il固定相还能基线分离其他异构体混样,如二氯苯异构体、二溴苯异构体、苯二酚异构体、苯二胺异构体、甲酚异构体、二甲苯酚异构体、烷基苯异构体、甲基萘/二甲基萘异构体等。
实施例2
三蝶烯二取代化合物tp-2il胍盐类离子液体具体制备步骤如下:
(1)将实施例1步骤(3)制备的tp-2br与四甲基-2-丁基胍按摩尔比1:2.2于乙腈中回流24h,旋蒸除去溶剂,粗产物经乙腈洗涤后得产物[tp-2r2]x2。
[tp-2r2]x2的核磁共振谱图的表征结果:1hnmr(400mhz,dmso,400mhz,298k),7.40(s,1h),6.99(s,1h),6.62(s,0h),5.83(s,0h),3.92(d,j=23.7hz,1h),3.17–3.05(m,1h),2.88(d,j=16.0hz,3h),1.74(d,j=6.0hz,1h),1.58–1.45(m,1h),1.43–1.19(m,3h),0.90(t,j=6.9hz,1h);13cnmr(101mhz,dmso)δ161.34,148.27,145.85,135.38–134.70,130.07,124.25,111.51,69.43,47.11,44.57,31.76,30.25–28.22,25.89–24.61,19.79,14.02。
(2)按照1:2.2的摩尔比将[tp-2r2]x2与阴离子交换剂lintf2在二氯甲烷中搅拌24h,用去离子水洗涤产物,直至洗涤液中滴加硝酸银不再产生沉淀,得到产物tp-2il。
将制得的tp-2il溶于二氯甲烷中,配制成浓度为0.25mg/ml的固定相溶液,制备石英毛细管色谱柱(5m)。采用该色谱柱分别对氯硝基苯异构体、二氯苯异构体进行分离,结果详见图6(a)和图6(b);其中,色谱条件:(a)120℃,(b)90℃,载气(氮气)流速1.0ml/min。这两种苯环位置异构体的间位和邻位有着相似的结构和相近的沸点,该固定相能基线分离这些难分离物质,表现出高选择性和分离能力。
实施例3
三蝶烯三取代化合物tp-3il苯并咪唑类离子液体的具体制备步骤如下:
(1)将2g三蝶烯加入70ml质量分数为68%的浓硝酸中,80℃下回流24h后,冷却至室温,在搅拌下倒入冷水中冷却析出,抽滤,粗产物用洗脱剂(v乙酸乙酯:v石油醚=4:1)在硅胶柱上分离纯化,制得2,6,14-三硝基三蝶烯。
2,6,14-三硝基三蝶烯的核磁共振氢谱的表征结果:1hnmr(400mhz,cdcl3):δ5.82(s,1h),5.83(s,1h),7.62-7.66(m,3h),8.04-8.07(m,3h),8.32-8.34(m,3h)。
(2)将1g2,6,14-三硝基三蝶烯、2gkbh4和0.2gpd/c(pd的负载量为5wt%)加入250ml三口瓶中,在氮气保护下,再加入40ml二氯甲烷和10ml无水甲醇,反应12h后,将反应物倒入装有硅藻土的砂芯漏斗中,滤去钯碳和未反应完的kbh4,滤液用水和饱和nacl溶液洗涤,收集有机相,用无水mgso4干燥,旋蒸处理后得到2,6,14-三氨基三蝶烯。
2,6,14-三氨基三蝶烯的核磁共振氢谱的表征结果:1hnmr(400mhz,cdcl3):δ3.49(s,6h),5.02(s,1h),5.04(s,1h),6.22-6.26(m,3h),6.70-6.72(m,3h),7.05-7.07(m,3h)。
(3)将0.55g2,6,14-三氨基三蝶烯溶于12ml质量浓度为10%的硫酸溶液中,充分搅拌使溶液由浑浊变为澄清,即将其转化成相应的盐,再置于冰盐浴中缓慢滴加6ml浓度为1mol/l的亚硝酸钠溶液,搅拌20min后将混合物再缓慢滴加到回流的35ml质量分数为50%的硫酸溶液中,继续回流2h后,冷却并用乙酸乙酯萃取,萃取物依次用水、饱和nacl溶液洗涤,随后用无水硫酸钠干燥,再旋蒸去除溶剂,粗产物用洗脱剂(v乙酸乙酯:v石油醚=1:4)在硅胶柱上分离提纯,得到产物2,6,14-三羟基三蝶烯。
2,6,14-三羟基三蝶烯的核磁共振氢谱的表征结果:1hnmr(400mhz,acetone-d6):δ5.25(s,2h),6.36-6.40(m,3h),6.90-6.92(m,3h),7.13-7.17(m,3h),8.01(s,3h)。
(4)将0.3g2,6,14-三羟基三蝶烯和0.6g碳酸钾溶于30ml乙腈中,氮气保护下,加入1.5g1,8-二溴辛烷,80℃下回流48h后,旋蒸除去溶剂,残余物先用水溶解,再用乙酸乙酯萃取,有机相用无水硫酸钠干燥,粗产物用洗脱剂(v乙酸乙酯:v石油醚=1:8)在硅胶柱上分离提纯,得到产物2,6,14-三(8-溴辛氧基)三蝶烯。
2,6,14-三(8-溴辛氧基)三蝶烯的核磁共振谱图的表征结果:1hnmr(400mhz,cdcl3):δ1.31-1.46(m,24h),1.67-1.74(m,6h),1.81-1.88(m,6h),3.38-3.41(t,6h),3.85-3.88(t,6h),5.19(s,1h),5.20(s,1h),6.43-6.46(m,3h),6.93-6.95(m,3h),7.19-7.22(m,3h);13cnmr(100mhz,cdcl3):δ13156.66,147.26,137.41,123.66,111.08,109.50,68.02,53.10,33.97,32.74,29.13,28.63,28.04,25.90。
(5)取1.18g苯并咪唑和1.12g氢氧化钾溶于二甲基亚砜中,搅拌1h后,再滴加2.49g1-溴正十二烷,室温搅拌24h后,再向反应体系加入50ml水,用氯仿萃取,有机相用水洗至中性,无水na2so4干燥,旋干溶剂真空烘干,得到淡黄色油状液体产物十二烷基苯并咪唑。
十二烷基苯并咪唑的核磁共振氢谱的表征结果:1hnmr(400mhz,cdcl3):δ0.88(t,3h),1.24-1.32(m,18h,),1.86-1.90(m,2h),4.16(t,2h),7.28-7.32(m,2h),7.39-7.41(m,1h),7.80-7.82(m,1h),7.89(s,1h)。
(6)取0.25g2,6,14-三(8-溴辛氧基)三蝶烯和0.26g(3.5eq.)十二烷基苯并咪唑,加入12ml乙腈,80℃下回流5天后,旋蒸除溶剂,并用乙醚洗涤,再置于50℃真空烘箱中烘干,制得产物[tp-3r2]x3。
[tp-3r2]x3的核磁共振谱图的表征结果:1hnmr(400mhz,dmso-d6):δ0.81-0.84(t,9h),1.19-1.29(m,78h),1.60(m,6h),1.90(m,12h),3.82-3.85(t,6h),4.48-4.51(t,12h),5.37(s,1h),5.38(s,1h),6.44-6.45(m,3h),6.98(m,3h),7.21-7.23(m,3h),7.67-7.69(m,6h),8.09-8.11(m,6h),9.93(s,3h);13cnmr(100mhz,dmso-d6):δ13156.21,147.80,142.18,137.76,131.15,126.58,123.82,113.80,110.97,109.46,67.56,46.71,31.33,29.05,28.95,25.76,22.13,13.00。esi-msm/z=498.3901(m+)。
(7)[tp-3r2]x3与的阴离子交换剂lintf2按照1:3.3的摩尔比溶于甲醇中,室温搅拌24h后,旋蒸除去溶剂,粗产物用水和二氯混合溶剂萃取,再用水洗至无br-存在,得到目标产物tp-3il。该目标产物的esi-msm/z=498.3901(m+),esi-msm/z=279.9170(m-)。
将所制备的tp-3il溶于二氯甲烷中,配制成浓度为0.25mg/ml的固定相溶液,并制备石英毛细管色谱柱(5m)。采用该色谱柱对癸烷、十一烷、苯乙醚、辛酮、苯甲醛、正辛醇、苯甲腈、苯胺、1,3-二溴苯、1,4-二甲氧基苯组成的混合样品进行分离,分离结果详见图7;其中,色谱条件:60℃~140℃,10℃/min,载气(氮气)流速1.0ml/min。由图可见,tp-3il固定相不仅高效分离各类组分,而且对易拖尾的胺类组分均得到对称的色谱峰,有利于样品组分色谱定性和定量分析测定。此外,tp-3il固定相还能基线分离各类异构体,如三氯苯异构体、三甲苯异构体、氯硝基苯异构体等。
实施例4
三蝶烯三取代化合物tp-3il咪唑类离子液体的具体制备步骤如下:
(1)取实施例3步骤(4)制备的2,6,14-三(8-溴辛氧基)三蝶烯0.12g和丁基咪唑0.068g(4eq.)于三口瓶中,再加入12ml乙腈,80℃下回流2天后,旋蒸除溶剂,并用乙醚洗洗涤,再置于50℃真空烘箱中烘干,制得产物[tp-3r2]x3。
[tp-3r]x3的核磁共振氢谱的表征结果:1hnmr(400mhz,dmso)δ0.86-0.90(t,9h),1.20-1.28(m,30h),1.63(m,6h),1.76-1.78(m,12h),3.86(t,6h),4.16(t,12h),5.38(s,1h),5.39(s,1h),6.46-6.48(m,3h),6.99(m,3h),7.22-7.24(m,3h),7.81(m,6h),9.28(s,3h),如图8所示。
(2)[tp-3r2]x3与阴离子交换剂lintf2按照1:3.3的摩尔比溶于甲醇中,室温搅拌24h后,旋蒸除去溶剂,粗产物用水和二氯混合溶剂萃取,再用水洗至无br-存在,得到目标产物tp-3il。该目标产物esi-msm/z=335.9146(m+),esi-msm/z=279.9159(m-)。
tp-3il的核磁共振谱图的表征结果:1hnmr(400mhz,dmso)δ0.89-0.90(t,9h),1.21-1.28(m,30h),1.63(m,6h),1.74-1.80(m,12h),3.86(t,6h),4.15(t,12h),5.37(m,2h),6.46-6.48(m,3h),6.99(m,3h),7.22-7.24(m,3h),7.78(m,6h)9.17(s,3h),如图9所示;13cnmr(100mhz,dmso-d6):δ13156.09,147.11,137.77,136.78,123.77,121.07,117.87,110.88,109.44,67.50,48.68,31.22,28.63,25.38,18.73,13.1。
将制备的tp-3il溶于二氯甲烷中,配制成浓度为0.25mg/ml的固定相溶液,并制备石英毛细管色谱柱(10m)。采用该色谱柱对丁基苯、正十二烷、壬酮、对二溴苯、癸酸甲酯、1,4-二甲氧基苯、萘、十一醇、对甲酚以及对氯苯酚组成的混合样品进行分离,分离结果详见图10;其中,色谱条件:60℃~140℃,10℃/min,载气(氮气)流速1.0ml/min。由图可见,tp-3il固定相色谱柱不仅高效分离各类组分,而且对易拖尾的醇类、酚类组分均得到对称的色谱峰,有利于提高样品组分色谱定性分析和定量分析测定结果的准确性。此外,该固定相色谱柱还能基线分离各类异构体,如二氯苯异构体、二溴苯异构体、苯二酚异构体、苯二胺异构体、甲胺异构体、二甲苯胺异构体、甲酚异构体、二甲苯酚异构体、丁二醇异构体等。
实施例5
三蝶烯一取代化合物tp-il咪唑类的离子液体具体制备步骤如下:
(1)将1g三蝶烯和1.04g无水氯化铝加入到30ml四氯乙烷中,在低温下(-15℃~-10℃)滴加0.31ml乙酰氯,搅拌40min后,再加入50ml冰盐酸,随后用二氯甲烷萃取得到紫色混合物,并真空干燥,粗产物采用洗脱剂(v乙酸乙酯:v石油醚=1:4)在硅胶柱上分离提纯,得到2-乙酰基三蝶烯。
(2)将0.71g2-乙酰基三蝶烯和0.89g溴化铜加入至25ml乙酸乙酯中,75℃下回流至不再产生气体和白色沉淀,滤去不溶杂质和未反应物,粗产物用洗脱剂(v乙酸乙酯:v石油醚=1:10)在硅胶柱上分离提纯,得到卤代2-乙酰基三蝶烯。
(3)将卤代2-乙酰基三蝶烯与丁基咪唑以1:1.1的摩尔比加入到无水乙腈中,80℃下回流反应24h,先置于0℃~4℃下冷却2h~3h,固体析出后,用二氯甲烷/石油醚(1:10,v/v)混合液多次洗涤,过滤析出的固体,得到产物[tp-r2]x。
[tp-r2]x的核磁共振谱图表征结果:1hnmr(400mhz,cdcl3)δ9.93(s,1h),7.93(s,1h),7.58(d,j=7.5hz,1h),7.33(d,j=7.5hz,1h),7.28–7.24(m,4h,arh),7.12(s,1h),6.94(s,1h),6.91–6.80(m,4h,arh),6.04(s,2h),5.45(s,1h),5.38(s,1h),3.88(t,j=7.2hz,2h),1.65-1.57(m,j=14.7,7.4hz,2h),1.19–1.11(m,2h),0.76(t,j=7.2hz,3h);13cnmr(101mhz,cdcl3)δ190.18,152.45,146.54,144.50,143.89,137.57,130.68,127.05,125.66,124.07,123.32,120.95,55.69,54.04,53.61,49.84,31.88,19.43,13.47。
(4)按照1:1.1的摩尔比将[tp-r2]x与lintf2溶于二氯甲烷中,室温下搅拌24h,有机相用去离子水多次洗涤,直至洗涤液中滴加硝酸银溶液后无白色沉淀生成,旋蒸除去溶剂,得到目标产物tp-il。
tp-il的核磁共振谱图表征结果:1hnmr(400mhz,cdcl3)δ8.74(s,1h),8.01(s,1h),7.66(d,j=7.6hz,1h),7.54–7.47(m,1h),7.46–7.36(m,4h),7.23(s,1h),7.18(s,1h),7.06–6.96(m,4h),5.69(s,2h),5.58(s,1h),5.51(s,1h),4.12(t,j=7.4hz,2h),1.92–1.77(m,2h),1.35(dt,j=14.8,7.4hz,2h),0.95(t,j=7.3hz,3h);13cnmr(101mhz,cdcl3)δ189.37,152.96,146.87,144.50,143.84,137.01,130.41,126.65,125.72,124.14,123.19,121.58,118.27,55.11,54.20,53.74,50.15,31.83,19.35,13.31。
将所制备的tp-il与二氯甲烷中配制成浓度为0.25mg/ml的固定相溶液,制备石英毛细管色谱柱(5m)。采用该色谱柱分别对庚烷异构体、苯二酚异构体进行分离,结果详见图11(a)和图11(b);其中,色谱条件:(a)120℃,载气(氮气)流速1.0ml/min;(b)90℃~160℃,10℃/min,载气(氮气)流速0.8ml/min。庚烷异构体结构相似,沸点也相似,tp-il固定相色谱柱通过三蝶烯部分的非极性作用,使其进行分离;该色谱柱还分离二甲苯酚异构体,其中2,3-二甲苯酚和3,5-二甲苯酚沸点极为相似,为难分离物质对。此外,tp-il固定相还能高效分离其他异构体混合物样品(如己烷异构体、辛烷异构体、硝基苯异构体、氯苯胺异构体、氯硝基苯异构体)以及不同类型组分混合物样品等。
综上所述,以上仅为本发明的较佳实施例而已,并非用于限定本发明的保护范围。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!