一种2-氯-3-氰基吡啶的绿色制备方法与流程
本发明属于有机中间体合成领域,具体涉及一种2-氯-3-氰基吡啶的绿色制备方法。
背景技术:
2-氯-3-氰基吡啶是医药工业中常见的重要中间体,其用途广泛,可以其为原料合成众多性能优良的药物,例如抗抑郁药米氮平,抗艾滋病药物奈韦拉平,同时还具有消炎止痛,治疗消耗综合症等功效,也可以其为原料合成农用化学品,以用于提高饲料蛋白的利用率,同时还可以用作杀虫剂和除草剂。
现有的2-氯-3-氰基吡啶合成路线有很多,其中一条是以3-氰基吡啶n-氧化物为原料,与氯化试剂进行反应,所用的氯化试剂大多以三氯氧磷、五氯化磷、二氯亚砜以及磺酰氯为主,主要存在以下不足:(1)使用pocl3作为氯化剂在回流条件下直接氯化,或增添pcl5加速反应,使用的氯化剂摩尔量不仅是原料摩尔量的3倍以上,并且产生了大量难以处理的含磷废水与废酸。(2)使用二氯亚砜及磺酰氯作为氯化试剂,会产生较多2位羟基取代的副产物以及大量的二氧化硫气体,具有较低反应选择性和反应收率。
专利jp8217753报道用双(三氯甲基)碳酸酯取代三氯氧磷做氯化试剂进行反应,收率仅为43%;专利cn101659637(a)在此基础上进行条件优化,报道收率在50%~85%;专利cn101941943(a)通过双(三氯甲基)碳酸酯与取代酰胺反应,先制得vilsmeier试剂,然后在高温条件下对底物进行氯化,报道收率基本在80%以上(纯度>98%)。按照专利cn101941943(a)报道的方式进行实验,未得到理想收率;在专利cn101659637(a)基础上,通过加入添加剂和相转移催化剂以及反应条件筛选,最终收率稳定提高至90%以上。且相比原有氯化方式,反应条件更加温和。
此制备方法采用绿色有机合成试剂双(三氯甲基)碳酸酯做氯化试剂,符合环保要求;而且工艺简单,操作方便,减少工业生产2-氯3-氰基吡啶及类似物带来的环境污染。
技术实现要素:
本发明克服了现有合成2-氯-3-氰基吡啶技术存在的上述缺陷,提出了一种环境较为友好的2-氯-3-氰基吡啶的制备方法,用双(三氯甲基)碳酸酯作为氯化试剂,与3-氰基吡啶n-氧化物反应,在添加剂以及相转移催化剂的作用下,大大提高反应收率,同时极大程度上降低原有技术路线对环境造成的污染。
本发明以3-氰基吡啶n-氧化物为原料,制备2-氯-3-氰基吡啶,包括以下步骤:
首先在有机溶剂中溶解3-氰基吡啶n-氧化物,于-5℃~10℃下加入有机碱;控制体系温度在-10~-5℃,加入添加剂与相转移催化剂;继续降温,在-15~-10℃下缓慢滴加溶于有机溶剂的双(三氯甲基)碳酸酯溶液,并于-15℃~60℃下反应2~16h。
上述反应过程如下式所示:
所述分离提纯过程可按照本领域常规方法进行,具体如下:反应结束后,加入少许水淬灭、分层萃取、有机层依次碱洗、水洗、萃取、脱色、干燥,浓缩得产物2-氯-3-氰基吡啶。
使用的溶剂为1,2-二氯乙烷、二氯甲烷、甲苯、1,4-二氧六环、乙腈、四氢呋喃、石油醚中的一种,优选为1,2-二氯乙烷。
使用的有机碱为三乙胺,n,n-二甲基甲酰胺、吡啶中的一种,优选为三乙胺。
使用的添加剂为氯化钠、氯化钾、氯化铝、亚硫酸钠、硫代硫酸钠、连二亚硫酸钠中的一种或多种,优选为氯化钠与亚硫酸钠的组合,此时,3-氰基吡啶n-氧化物与氯化钠的摩尔比为1:0.5~1;3-氰基吡啶n-氧化物与亚硫酸钠的摩尔比为1:0.01~0.05,此时,反应收率可以稳定地达到90%以上。
为得到较多目标产品,避免原料及氯化试剂浪费,作为优选,所用3-氰基吡啶n-氧化物与双(三氯甲基)碳酸酯及有机碱的摩尔比为1:0.4~1:1.5~3.5,所用溶剂与3-氰基吡啶n-氧化物的重量比为5~20:1。
本发明中,所述的3-氰基吡啶n-氧化物和相转移催化剂的摩尔比为1:0.01~0.04
本发明与现有技术相比具有以下优点:
(1)双(三氯甲基)碳酸酯是一种较安全的氯化试剂,利用其代替二氯亚砜、磺酰氯,三氯氧磷、五氯化磷等可避免二氧化硫、含磷废水等高污染物的生成,对环境影响小。
(2)工艺简单,反应条件相对于已有方法较温和,且后处理方便,最终产品纯度和收率高,适合工业化生产。
附图说明
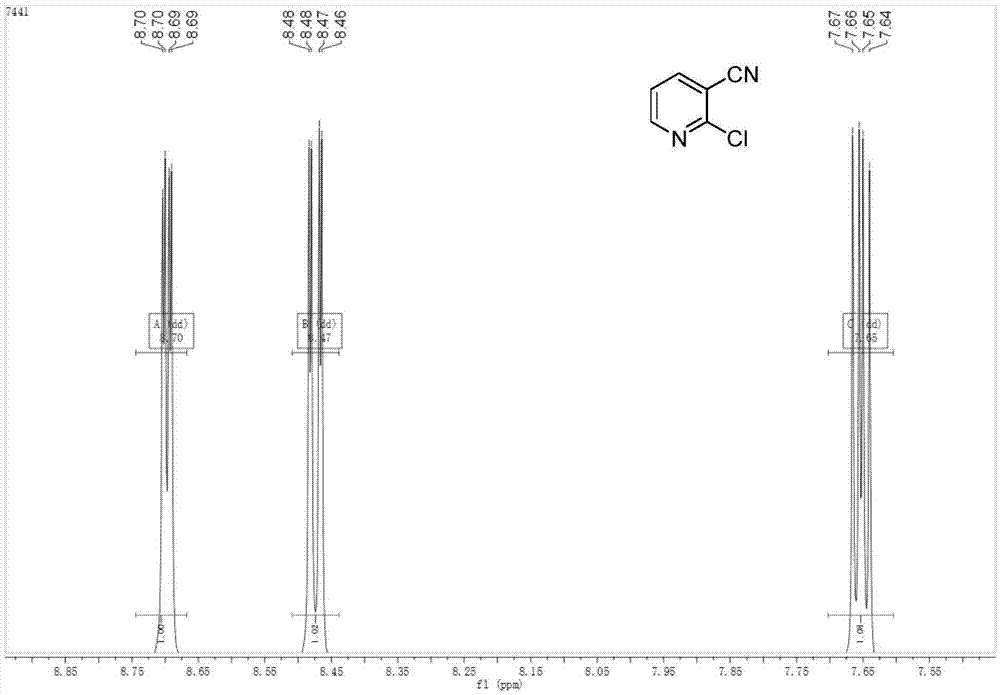
图1为本发明具体实施例1中所得产品2-氯-3氰基吡啶的氢核磁谱图(1h-nmr),核磁数据如下:1hnmr(500mhz,dmso)δ8.70(dd,j=4.9,1.9hz,1h),8.47(dd,j=7.8,1.9hz,1h),7.65(dd,j=7.8,4.9hz,1h).
具体实施方式
实施例1
取一干燥的四口烧瓶,搭好尾气吸收装置,加入15g(125mmol)3-氰基吡啶n-氧化物及110ml1,2-二氯乙烷,在0℃的低温条件下搅拌成均质溶液,当t=-5~0℃时,加入25.30g(250mmol)三乙胺,继续降温,加入3.50g(62.5mmol)氯化钠,0.78g(6.25mmol)亚硫酸钠,1.13g(5mmol)苄基三乙基氯化铵,继续冷却至-15℃,开始缓慢滴加60ml含18.52g(62.5mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,于该温度下保温反应16小时,反应完毕,滴加40ml去离子水搅拌淬灭。分层,有机相用60ml10%的氢氧化钠溶液洗涤,再用60ml去离子水洗涤,最后用(60ml*2)1,2-二氯乙烷萃取水相,合并有机层,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机相用无水硫酸镁干燥后,脱除溶剂,粗产物打浆,烘干后,得16.30g红褐色产物,收率94.15%。
实施例2
取一干燥的四口烧瓶,搭好尾气吸收装置,加入15g(125mmol)3-氰基吡啶n-氧化物及110ml1,2-二氯乙烷,在0℃的低温条件下搅拌成均质溶液,当t=-5~0℃时,加入31.63g(312.5mmol)三乙胺,继续降温,加入1.83g(31.25mmol)氯化钠,0.78g(6.25mmol)亚硫酸钠,1.13g(5mmol)苄基三乙基氯化铵,继续冷却至-15℃,开始缓慢滴加60ml含18.52g(62.5mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,于该温度下保温反应16小时,反应完毕,滴加40ml去离子水搅拌淬灭。分层,有机相用60ml10%的氢氧化钠溶液洗涤,再用60ml去离子水洗涤,最后用(60ml*2)1,2-二氯乙烷萃取水相,合并有机层,加入活性炭于60℃下回流脱色一小时,脱色完毕,过滤,有机相用无水硫酸镁干燥后,脱除溶剂,粗产物打浆,烘干后,得13.74g红褐色产物,收率79.36%。
实施例3
取一干燥的四口烧瓶,搭好尾气吸收装置,加入15g(125mmol)3-氰基吡啶n-氧化物及110ml1,2-二氯乙烷,在0℃的低温条件下搅拌成均质溶液,当t=-5~0℃时,加入31.63g(312.5mmol)三乙胺,继续降温,加入3.50g(62.5mmol)氯化钠,0.78g(6.25mmol)亚硫酸钠,1.13g(5mmol)苄基三乙基氯化铵,继续冷却至-15℃,开始缓慢滴加60ml含18.52g(62.5mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,于该温度下保温反应15小时,反应完毕,滴加40ml去离子水搅拌淬灭。分层,有机相用60ml10%的氢氧化钠溶液洗涤,再用60ml去离子水洗涤,最后用(60ml*2)1,2-二氯乙烷萃取水相,合并有机层,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,用无水硫酸镁干燥后,脱除溶剂,粗产物打浆,烘干后,得14.76g红褐色产物,收率85.26%。
实施例4
取一干燥的四口烧瓶,搭好尾气吸收装置,加入15g(125mmol)3-氰基吡啶n-氧化物及110ml1,2-二氯乙烷,在0℃的低温条件下搅拌成均质溶液,当t=-5~0℃时,加入37.95g(375mmol)三乙胺,继续降温,加入1.83g(31.25mmol)氯化钠,0.78g(6.25mmol)亚硫酸钠,1.13g(5mmol)苄基三乙基氯化铵,继续冷却至-15℃,开始缓慢滴加100ml含29.63g(100mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,于该温度下保温反应4小时,反应完毕,滴加40ml去离子水搅拌淬灭。分层,有机相用60ml10%的氢氧化钠溶液洗涤,再用60ml去离子水洗涤,最后用(60ml*2)1,2-二氯乙烷萃取水相,合并有机层,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机相用无水硫酸镁干燥后,脱除溶剂,粗产物打浆,烘干后,得14.85g红褐色产物,收率85.76%。
实施例5
取一干燥的四口烧瓶,搭好尾气吸收装置,加入15g(125mmol)3-氰基吡啶n-氧化物及110ml1,2-二氯乙烷,在0℃的低温条件下搅拌成均质溶液,当t=-5~0℃时,加入37.95g(375mmol)三乙胺,继续降温,加入1.83g(31.25mmol)氯化钠,0.78g(6.25mmol)亚硫酸钠,1.13g(5mmol)苄基三乙基氯化铵,继续冷却至-15℃,开始缓慢滴加120ml含37.03g(125mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,于该温度下保温反应3小时,反应完毕,滴加40ml去离子水搅拌淬灭。分层,有机相用60ml10%的氢氧化钠溶液洗涤,再用60ml去离子水洗涤,最后用(60ml*2)1,2-二氯乙烷萃取水相,合并有机层,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机相用无水硫酸镁干燥后,脱除溶剂,粗产物打浆,烘干后,得13.52g红褐色产物,收率78.10%。
实施例6
取一干燥的四口烧瓶,搭好尾气吸收装置,加入15g(125mmol)3-氰基吡啶n-氧化物及110ml1,2-二氯乙烷,在0℃的低温条件下搅拌成均质溶液,当t=-5~0℃时,加入37.95g(375mmol)三乙胺,继续降温,加入1.83g(31.25mmol)氯化钠,0.78g(6.25mmol)亚硫酸钠,1.13g(5mmol)苄基三乙基氯化铵,继续冷却至-15℃,开始缓慢滴加120ml含37.03g(125mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,升温至60℃,并在该温度下保温反应10小时,反应完毕,滴加40ml去离子水搅拌淬灭。分层,有机相用60ml10%的氢氧化钠溶液洗涤,再用60ml去离子水洗涤,最后用(60ml*2)1,2-二氯乙烷萃取水相,合并有机层,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机层用无水硫酸镁干燥后,脱除溶剂,粗产物打浆,烘干后,得14.58g红黑色产物,收率84.22%。
实施例7
取一干燥的四口烧瓶,搭好尾气吸收装置,加入2.40g(20mmol)3-氰基吡啶n-氧化物,7.00g(23.4mmol)双(三氯甲基)碳酸酯及25ml石油醚,并于0℃下滴加30ml含7.08g(70mmol)三乙胺的石油醚溶液,滴加完毕,升温至60℃,并在该温度下保温反应4小时,反应完毕,冷却至室温,依次用20ml去离子水洗涤,再用20ml10%的碳酸氢钠溶液洗涤。分层,收集有机相,加入活性碳,于60℃下回流脱色一小时,脱色完毕,过滤,有机层用无水硫酸镁干燥后,脱除溶剂,得1.46g砖红色产物,收率52.3%。
实施例8
取一干燥的四口烧瓶,搭好尾气吸收装置,加入6g(50mmol)3-氰基吡啶n-氧化物,7.60g(75mmol)三乙胺及40ml1,2-二氯乙烷,并于-15℃下滴加25ml含7.40g(25mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,并在该温度下保温反应2小时,反应完毕,升温至室温,加少许水淬灭后,分层,依次用20ml10%的碳酸氢钠溶液洗涤,再用20ml去离子水洗涤。分层,收集有机相,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机层用无水硫酸镁干燥后,脱除溶剂,得5.07g砖红色产物,收率73.21%。
实施例9
取一干燥的四口烧瓶,搭好尾气吸收装置,加入12g(100mmol)3-氰基吡啶n-氧化物,20.24g(200mmol)三乙胺及100ml1,2-二氯乙烷,并于-10℃下滴加60ml含17.78g(60mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,并在该温度下保温反应4小时,反应完毕,升温至室温,加少许水淬灭后,分层,依次用25ml10%的碳酸氢钠溶液洗涤,再用40ml去离子水洗涤。分层,收集有机相,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机层用无水硫酸镁干燥后,脱除溶剂,得11.83g红色产物,收率85.40%。
实施例10
取一干燥的四口烧瓶,搭好尾气吸收装置,加入6g(50mmol)3-氰基吡啶n-氧化物,7.60g(75mmol)三乙胺及40ml二氯甲烷,并于-15℃下滴加25ml含7.40g(25mmol)双(三氯甲基)碳酸酯的二氯甲烷溶液,滴加完毕,并在该温度下保温反应2小时,反应完毕,升温至室温,加少许水淬灭后,分层,依次用20ml10%的碳酸氢钠溶液洗涤,再用20ml去离子水洗涤。分层,收集有机相,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机层用无水硫酸镁干燥后,脱除溶剂,得4.87g砖红色产物,收率70.32%。
实施例11
取一干燥的四口烧瓶,搭好尾气吸收装置,加入6g(50mmol)3-氰基吡啶n-氧化物,7.60g(75mmol)三乙胺及40ml四氢呋喃,并于-15℃下滴加25ml含7.40g(25mmol)双(三氯甲基)碳酸酯的四氢呋喃溶液,滴加完毕,并在该温度下保温反应2小时,反应完毕,升温至室温,加少许水淬灭后,分层,依次用20ml10%的碳酸氢钠溶液洗涤,再用20ml去离子水洗涤。分层,收集有机相,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,用无水硫酸镁干燥后,脱除溶剂,得3.58g砖红色产物,收率51.70%。
实施例12
取一干燥的四口烧瓶,搭好尾气吸收装置,加入6g(50mmol)3-氰基吡啶n-氧化物,7.60g(75mmol)三乙胺及40ml甲苯,并于-15℃下滴加25ml含7.40g(25mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,升温至60℃,并在该温度下保温反应2小时,反应完毕,降温至室温,加少许水淬灭后,分层,依次用20ml10%的碳酸氢钠溶液洗涤,再用20ml去离子水洗涤。分层,收集有机相,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机相用无水硫酸镁干燥后,脱除溶剂,得4.52g红色产物,收率65.27%。
实施例13
取一干燥的四口烧瓶,搭好尾气吸收装置,加入6g(50mmol)3-氰基吡啶n-氧化物,9.93g(75mmol)吡啶及40ml1,2-二氯乙烷,并于-15℃下滴加25ml含7.40g(25mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,并在该温度下保温反应2小时,反应完毕,升温至室温,加少许水淬灭后,分层,依次用20ml10%的氢氧化钠溶液洗涤,再用20ml去离子水洗涤。分层,收集有机相,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机相用无水硫酸镁干燥后,脱除溶剂,得2.98g红褐色产物,收率43.03%。
实施例14
取一干燥的四口烧瓶,搭好尾气吸收装置,加入6g(50mmol)3-氰基吡啶n-氧化物,5.06g(50mmol)三乙胺及40ml1,2-二氯乙烷,当温度降至0℃后,加入0.56g(2mmol)四丁基氯化铵及5.85g(100mmol)氯化钠,并于-10℃下滴加25ml含7.41g(25mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,并在该温度下保温反应2小时,之后升温至60℃反应2小时,反应结束,降至室温,加少许水淬灭后,分层,依次用20ml10%的氢氧化钠溶液洗涤,再用20ml去离子水洗涤。分层,收集有机相,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机相用无水硫酸镁干燥后,脱除溶剂,得4.47g淡红褐色产物,收率64.52%。
实施例15
取一干燥的四口烧瓶,搭好尾气吸收装置,加入6g(50mmol)3-氰基吡啶n-氧化物,12.65g(125mmol)三乙胺及40ml1,2-二氯乙烷,当温度降至0℃后,加入1.09g(8mmol)三氯化铝,并于-10℃下滴加40ml含11.85g(40mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,并在该温度下保温反应半小时小时,之后升温至40℃反应5小时,反应结束,降至室温,加少许水淬灭后,分层,依次用20ml10%的碳酸氢钠溶液洗涤,再用20ml去离子水洗涤。分层,收集有机相,并用(25ml*2)1,2-二氯乙烷萃取水相,合并有机相,加入活性炭于60℃下回流脱色一小时,脱色完毕,过滤,有机相用无水硫酸镁干燥后,脱除溶剂,得5.14g白褐色产物,收率74.17%。
实施例16
取一干燥的四口烧瓶,搭好尾气吸收装置,加入6g(50mmol)3-氰基吡啶n-氧化物,12.65g(125mmol)三乙胺及50ml1,2-二氯乙烷,当温度降至0℃后,加入0.63g(2mmol)苄基三乙基氯化铵及2.93g(50mmol)氯化钠,并于-10℃下滴加50ml含14.84g(50mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,并在该温度下保温反应2小时,反应结束,加入40ml去离子水淬灭,分层,有机相依次用20ml10%的氢氧化钠溶液洗涤,再用20ml去离子水洗涤。分层,收集有机相,并用(25ml*2)1,2-二氯乙烷萃取水相,合并有机相,加入活性炭于60℃下回流脱色一小时,脱色完毕,过滤,用无水硫酸镁干燥后,脱除溶剂,得6.33g酒红色产物,收率91.41%。
实施例17
取一干燥的四口烧瓶,搭好尾气吸收装置,加入12g(100mmol)3-氰基吡啶n-氧化物,25.30g(250mmol)三乙胺及90ml1,2-二氯乙烷,当温度降至0℃后,加入2.13g(16mmol)三氯化铝,并于-10℃下滴加90ml含29.68g(100mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,并在该温度下保温反应2小时,反应结束,加入80ml去离子水淬灭,分层,有机相依次用100ml10%的氢氧化钠溶液洗涤,再用100ml去离子水洗涤。分层,收集有机相,并用(60ml*2)1,2-二氯乙烷萃取水相,合并有机相,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机相用无水硫酸镁干燥后,脱除溶剂,得11.60g红黑色产物,收率74.11%。
实施例18
取一干燥的四口烧瓶,搭好尾气吸收装置,加入6g(50mmol)3-氰基吡啶n-氧化物,7.59g(75mmol)三乙胺及50ml1,2-二氯乙烷,当温度降至0℃后,加入1.26g(10mmol)亚硫酸钠,并于-10℃下滴加50ml含14.81g(50mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,并在该温度下保温反应4小时,反应结束,加入50ml去离子水淬灭,分层,有机相依次用20ml10%的氢氧化钠溶液洗涤,再用20ml去离子水洗涤。分层,收集有机相,并用(25ml*2)1,2-二氯乙烷萃取水相,合并有机相,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机层用无水硫酸镁干燥后,脱除溶剂,得4.54g白褐色产物,收率65.56%。
实施例19
取一干燥的四口烧瓶,搭好尾气吸收装置,加入6g(50mmol)3-氰基吡啶n-氧化物,12.65g(125mmol)三乙胺及50ml1,2-二氯乙烷,当温度降至0℃后,加入0.63g(5mmol)亚硫酸钠及0.67g(5mmol)三氯化铝,并于-10℃下滴加50ml含14.81g(50mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,并在该温度下保温反应3小时,反应结束,加入40ml去离子水淬灭,分层,有机相依次用40ml10%的氢氧化钠溶液洗涤,再用40ml去离子水洗涤。分层,收集有机相,并用(30ml*2)1,2-二氯乙烷萃取水相,合并有机相,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机相用无水硫酸镁干燥后,脱除溶剂,得4.99g砖红色产物,收率72.12%。
实施例20
取一干燥的四口烧瓶,搭好尾气吸收装置,加入12g(100mmol)3-氰基吡啶n-氧化物,25.30g(250mmol)三乙胺及90ml1,2-二氯乙烷,当温度降至0℃后,加入1.29g(4mmol)苄基三乙基氯化铵及5.84g(100mmol)氯化钠,并于-5℃下滴加100ml含29.63g(100mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,并在该温度下保温反应3小时,反应结束,加入40ml去离子水淬灭,分层,有机相依次用20ml10%的氢氧化钠溶液洗涤,再用20ml去离子水洗涤。分层,收集有机相,并用(25ml*2)1,2-二氯乙烷萃取水相,合并有机相,加入活性炭,于60℃下回流脱色一小时,脱色完毕,过滤,有机相用无水硫酸镁干燥后,脱除溶剂,得11.92g褐色产物,收率86.3%。
实施例21
取一干燥的四口烧瓶,搭好尾气吸收装置,加入6g(50mmol)3-氰基吡啶n-氧化物,12.65g(125mmol)三乙胺及50ml1,2-二氯乙烷,当温度降至0℃后,加入0.63g(2mmol)苄基三乙基氯化铵及0.63g(5mmol)亚硫酸钠,并于-15℃下滴加50ml含14.84g(50mmol)双(三氯甲基)碳酸酯的1,2-二氯乙烷溶液,滴加完毕,并在该温度下保温反应5小时,反应结束,加入40ml去离子水淬灭,分层,有机相依次用30ml10%的氢氧化钠溶液洗涤,再用30ml去离子水洗涤。分层,收集有机相,并用(40ml*2)1,2-二氯乙烷萃取水相,合并有机相,加入活性炭于60℃下回流脱色一小时,脱色完毕,过滤,有机相用无水硫酸镁干燥后,脱除溶剂,得5.83g褐色产物,收率84.20%。
实施例1~21中所得目标产品经核磁数据、熔点数据检测,与已报道文献中2-氯-3-氰基吡啶基本一致。
熔点:106.0℃-106.3℃。
- 还没有人留言评论。精彩留言会获得点赞!