含铱氮双重键的三价铱亚胺配合物、制备方法及其应用与流程
本发明属于合成化学技术领域,具体涉及含铱氮双重键的三价铱亚胺化合物、制备方法及其应用。
背景技术:
直链胺类化合物可通过各种已知反应转化成重要的化工中间体,因而其在农药、化妆品、医药用品的合成中有着非常广泛的应用。因此,胺类化合物的合成一直是化学领域研究的重点之一。传统的合成方法如硝基或氰基化合物的还原、酰胺的霍夫曼降解等从操作的简便性、原料的易得性、反应的选择性和原子经济性上看都有着不少的缺点。而氢胺化反应避免了盐类(如卤盐等)等副产物的产生,理论上两种原料中的每一个原子都出现在产物中。因而与其它各种胺化反应相比,氢胺化反应具有非常高的原子经济性,更加符合绿色化学的理念,从而被广泛地应用于各种天然产物或药物中间体的合成中,因此烯烃的反马氏氢胺化反应是合成高价值直链型胺类化合物的最优方法。
技术实现要素:
为了克服现有技术的不足,本发明的目的在于提出一种含铱氮双重键的三价铱亚胺化合物、制备方法及其应用。本发明制备方法简单绿色,得到的三价铱配合物能高效催化烯烃的反马氏氢胺化反应合成直链胺类化合物,可催化底物种类较多,普适性好,原料简单易得,反应条件温和,对于不同电子效应和空间位阻效应的底物均具有较高的催化活性,催化效率高,原子经济性高,成本较低且产物易于分离,不会产生大量废渣。
本发明的技术方案具体介绍如下。
本发明提供一种含铱氮双重键的三价铱亚胺化合物,其用作高效催化烯烃的反马氏氢胺化反应合成直链胺类化合物的催化剂,具有如下所示结构:
本发明还提供一种含铱氮双重键的三价铱亚胺化合物的制备方法,具体步骤如下:
在-78℃的温度下,将n-buli的正己烷溶液滴加到含苯基二吡咯的四氢呋喃溶液中,滴加结束后继续搅拌30-60分钟,缓慢升至室温后继续反应30-60分钟后加入一价铱前驱体环辛二烯氯化铱二聚体[(cod)ircl]2,继续在室温下反应3.0~6.0小时;然后将叠氮苯phn3加入反应体系反应,室温下反应2.0~3.0小时,反应结束后,静置过滤,减压抽干溶剂,得到的粗产物进行柱层析分离得到暗红色的目标产物。
本发明中,n-buli、苯基二吡咯、[(cod)ircl]2和叠氮苯phn3的摩尔比为(1.2~2.5):1:0.5:1.5。
本发明中,加入环辛二烯氯化铱二聚体[(cod)ircl]2,反应时间为3-6h。
本发明中,柱层析分离用到的洗脱剂是体积比在10:1~6:1之间的石油醚和二氯甲烷组成的混合溶剂。
本发明进一步提供一种上述的含铱氮双重键的三价铱亚胺化合物在催化烯烃的反马氏氢胺化反应合成直链胺类化合物中的应用。
本发明中,烯烃反马氏氢胺化反应中采用一级胺为原料。优选的,采用未取代苯胺或者c1~c5烷基、c1~c5烷氧基、硝基、卤素取代的苯胺为原料。
本发明中,烯烃反马氏氢胺化反应中的烯烃为苯乙烯。
本发明中,具体应用方法如下:按照原料一级胺和铱亚胺配合物的投料摩尔比为3000:1~1000:1,胺与苯乙烯的摩尔比为1:1,向一级胺和苯乙烯中加入含铱亚胺配合物的甲苯溶液,反应温度为25~60℃,反应时间为80~360分钟,反应结束后浓缩反应液经硅胶柱层析分离,得到直链胺类化合物。
和现有技术相比,本发明的有益效果在于,
(1)本发明中含铱氮双重键的三价铱亚胺化合物制备方法简单,具有较高收率;
(2)本发明中含铱氮双重键的三价铱亚胺化合物具有较高的热稳定性,在300℃下依然不分解;
(3)本发明的三价铱亚胺化合物,在温和(25-60℃)的条件下具有较高的烯烃氢胺化反应的催化活性,收率高(85%~97%),且区域选择性好,均为反马氏直链胺基产物。
附图说明
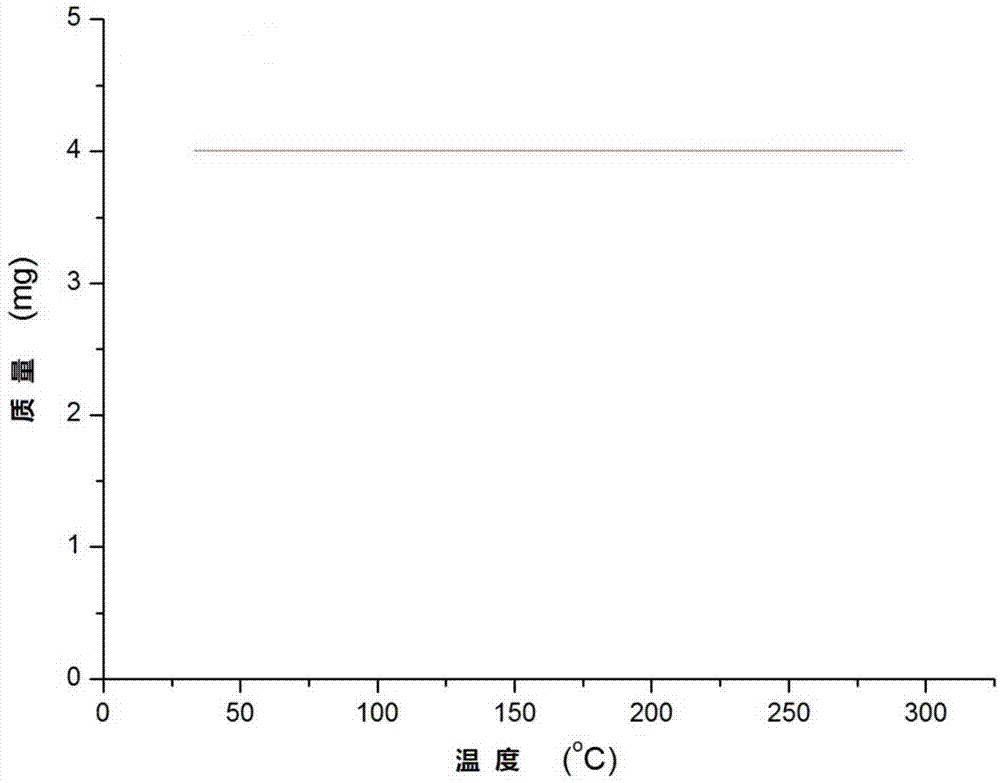
图1为实施例1获得的三价铱亚胺化合物的热重曲线。
具体实施方式
下面通过实施例进一步具体描述本发明,本发明并不局限于下述实施例。
实施例1:含铱氮双重键的三价铱亚胺化合物的合成
–78℃下,将n-buli(1.6m)的正己烷溶液(0.50ml,0.8mmol)缓慢滴加到含苯基二吡咯c15h14n2(127.0mg,0.57mmol)的四氢呋喃溶液中,在该温度下搅拌1小时,缓慢升至室温后继续反应1小时后加入一价铱前驱体环辛二烯氯化铱二聚体[(cod)ircl]2(188.0mg,0.28mmol),继续在室温下反应3小时。然后将叠氮苯phn3(50.0mg,0.42mmol)加入反应体系另外再反应3小时。反应结束后,静置过滤,减压抽干溶剂,得到的粗产物进行柱层析分离(石油醚/二氯甲烷(v/v)=8:1)得到暗红色的目标产物三价铱亚胺配合物c21h18irn3(115mg,产率81%)。
1hnmr(400mhz,cdcl3,25℃):δ=8.67-8.59(m,3h,),8.45-8.36(m,2h),8.22-8.12(m,5h,ph),7.69-7.54(m,5h,ph),4.36(d,2h),3.65(s,1h).元素分析理论值:c49.98,h3.60,n8.33;实验值:c49.86,h3.71,n8.25。
将三价铱亚胺配合物在甲苯溶液中加热回流三小时,反应冷却抽干溶剂,得到的固体进行核磁表征,各核磁信号无变化。此外,称取4.0mg化合物进行热重实验,结果显示在300℃高温下化合物依然稳定(热重曲线如图1所示)。
实施例2:三价铱亚胺配合物催化烯烃的反马氏氢胺化反应
采用实施例1制备的催化剂催化烯烃的反马氏氢胺化反应:向苯乙烯(1mmol,104mg)和苯胺(1mmol,93mg)中加入含有三价铱亚胺配合物(0.001mmol,5.0mg)的甲苯溶液,反应温度25℃,反应时间为90分钟,结束后浓缩反应液直接经硅胶柱层析分离,干燥至质量不变,得到对应的胺类化合物c14h15n(177mg,产率88%),元素分析:c85.24、h7.66、n7.10(理论);c85.33、h7.69、n7.12(实际)。
实施例3:三价铱亚胺配合物催化烯烃的反马氏氢胺化反应
采用实施例1制备的催化剂催化烯烃的反马氏氢胺化反应:向苯乙烯(1mmol,104mg)和苯胺(1mmol,93mg)中加入含有三价铱亚胺配合物(0.002mmol,10.0mg)的甲苯溶液,反应温度40℃,反应时间为80分钟,结束后浓缩反应液直接经硅胶柱层析分离,干燥至质量不变,得到对应的胺类化合物c15h17n(190mg,产率90%),元素分析:c85.26、h8.11、n6.63(理论);c85.19、h8.19、n6.73(实际)。
实施例4:三价铱亚胺配合物催化烯烃的反马氏氢胺化反应
采用实施例1制备的催化剂催化烯烃的反马氏氢胺化反应:向苯乙烯(1mmol,104mg)和苯胺(1mmol,93mg)中加入含有三价铱亚胺配合物(0.002mmol,10.0mg)的甲苯溶液,反应温度25℃,反应时间为120分钟,结束后浓缩反应液直接经硅胶柱层析分离,干燥至质量不变,得到对应的胺类化合物c15h17n(192mg,产率91%),元素分析:c85.26、h8.11、n6.63(理论);c85.13、h8.16、n6.55(实际)。
实施例5:三价铱亚胺配合物催化烯烃的反马氏氢胺化反应
采用实施例1制备的催化剂催化烯烃的反马氏氢胺化反应:向苯乙烯(1mmol,104mg)和苯胺(1mmol,93mg)中加入含有三价铱亚胺配合物(0.001mmol,5.0mg)的甲苯溶液,反应温度50℃,反应时间为100分钟,结束后浓缩反应液直接经硅胶柱层析分离,干燥至质量不变,得到对应的胺类化合物c15h17no(211mg,产率93%),元素分析:c79.26、h7.54、n6.16(理论);c79.11、h7.36、n6.25(实际)。
实施例6:三价铱亚胺配合物催化烯烃的反马氏氢胺化反应
采用实施例1制备的催化剂催化烯烃的反马氏氢胺化反应:向苯乙烯(1mmol,104mg)和苯胺(1mmol,93mg)中加入含有三价铱亚胺配合物(0.002mmol,10.0mg)的甲苯溶液,反应温度60℃,反应时间为240分钟,结束后浓缩反应液直接经硅胶柱层析分离,干燥至质量不变,得到对应的胺类化合物c16h19n(198mg,产率88%),元素分析:c85.28、h8.50、n6.22(理论);c85.41、h8.36、n6.35(实际)。
实施例7:三价铱亚胺配合物催化烯烃的反马氏氢胺化反应
采用实施例1制备的催化剂催化烯烃的反马氏氢胺化反应:向苯乙烯(1mmol,104mg)和苯胺(1mmol,93mg)中加入含有三价铱亚胺配合物(0.003mmol,15.0mg)的甲苯溶液,反应温度60℃,反应时间为200分钟,结束后浓缩反应液直接经硅胶柱层析分离,干燥至质量不变,得到对应的胺类化合物c14h14cln(224mg,产率97%),元素分析:c72.57、h6.09、n6.04(理论);c72.71、h6.16、n6.20(实际)。
实施例8:三价铱亚胺配合物催化烯烃的反马氏氢胺化反应
采用实施例1制备的催化剂催化烯烃的反马氏氢胺化反应:向苯乙烯(1mmol,104mg)和苯胺(1mmol,93mg)中加入含有三价铱亚胺配合物(0.003mmol,15.0mg)的甲苯溶液,反应温度60℃,反应时间为360分钟,结束后浓缩反应液直接经硅胶柱层析分离,干燥至质量不变,得到对应的胺类化合物c14h14n2o2(206mg,产率85%),元素分析:c69.41、h5.82、n11.56(理论);c69.55、h6.01、n11.36(实际)。
- 还没有人留言评论。精彩留言会获得点赞!