含氮盘状离子液晶化合物及其制备方法与流程
本发明涉及一种含氮盘状离子液晶化合物及其制备方法。
背景技术:
离子液晶(ilc)是结合了液晶和离子液体部分性质的功能材料,仅由阳离子和阴离子组成。这样独特的性质使得离子液晶在离子传导材料,有序反应介质,功能纳米材料等方面有广泛的应用前景。离子液晶可以通过改变阴阳离子的种类和大小来调节离子液晶的性质(如液晶范围,发光性等)以适应不同功能的需求。在1938年首次报道了ilc,而到目前为止,大多数ilc的研究主要集中在棒状ilc上,而具有柱状中间相的盘状ilc的有关报道还较少。其原因可能是盘状离子液晶复杂的合成、表征和繁琐的纯化步骤。近年来,盘状离子液晶的研究主要集中在具有软间隔基连接的吡啶鎓或咪唑鎓单元的苯并菲核上,它们显示出有限的化学结构变化和狭窄的中间相范围。因此,亟待通过分子设计、合成新的液晶温度应用范围宽,发光性能好的盘状离子液晶材料。
技术实现要素:
本发明的发明目的就是提供一种新型的盘状离子液晶材料:含氮盘状离子液晶化合物,该类化合物在较宽的温度范围内呈现柱状液晶相,且表现出良好的光学性质。
本发明实现其第一个发明目的,所采用的技术方案是:一种含氮盘状离子液晶化合物,其特征在于,所述化合物具有如下通式i的结构,其中r1=cnh2n+1,n=5,6,7,r2=cnh2n+1,n=1-6,x为无机阴离子或者有机阴离子,如无机阴离子:f-,cl-,br-,i-,no3-,bf4-,pf6-,so4-,h2po4-,clo4-,scn-,asf6-,有机阴离子:
其中,无机阴离子优选:br-,i-,no3-,bf4-,pf6-,有机阴离子优选:
与现有技术相比,本发明的通式为i的含氮盘状离子液晶化合物的有益效果是:
一、化学结构变化多样,可通过调节—r1、—r2的长度、阴阳离子的种类和大小来调节液晶性质,以适应不同功能的需求;
二、含氮盘状离子液晶表现出与一般盘状离子液晶不同的液晶行为,如易于呈现室温液晶,具有较宽温度应用范围等。实验证明,本发明的物质在常温下呈液晶状态,其相变温度为78℃以上,是一种在较宽的温度范围内呈现液晶相的新型液晶材料,具有良好的应用价值和前景;
三、含氮盘状离子液晶表现出良好的发光性质,在thf溶液和薄膜中发黄光,可应用于发光材料。
本发明的第二个发明目的是提供上述的通式为i的含氮盘状离子液晶化合物的制备方法。
本发明实现其第二个发明目的,所采用的技术方案是:一种上述的通式为i的含氮盘状离子液晶化合物的制备方法,其步骤是:
a、suzuki偶联反应
将1摩尔份的3,4-二溴吡啶、2.5摩尔份的芳基硼酸、10摩尔份的碳酸钾、0.25摩尔份的四(三苯基膦)合鈀加入到thf和h2o的混合溶液中,在氩气保护下,70℃搅拌反应24-36小时,反应结束后用二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯,得到的粘稠状化合物即为产物;
b、分子内芳烃氧化偶联反应
将a步得到的1摩尔份产物溶解在二氯甲烷中,缓慢加入溶解在硝基甲烷中的3摩尔份的三氯化铁溶液,滴加完后,室温搅拌反应2-3小时,反应过程中用薄层色谱跟踪至反应物反应完全,然后加入甲醇淬灭反应,用饱和食盐水洗涤,调节ph至碱性,二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯,石油醚重结晶,得到的白色固体化合物即为含氮苯并菲;
c、烷基化反应
将1摩尔份的含氮苯并菲溶于甲苯中,加入5摩尔份的卤代烃,40-80℃搅拌反应24-36小时,反应结束后,蒸去甲苯,将剩余物用硅胶柱层析分离提纯,用无水乙醇/无水甲醇重结晶,过滤,真空干燥,得到的黄色固体化合物即为烷基化产物;
d、离子交换反应(复分解反应)
将c步得到的1摩尔份的烷基化产物溶解在二氯甲烷中,加入3-5摩尔份的无机盐或有机盐,室温下搅拌反应3-5小时,反应结束后,用蒸馏水洗涤有机相3次,无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯,重结晶,过滤,真空干燥,得到的黄色固体化合物即为含氮盘状离子液晶化合物。
这种制备方法合成路线较短,而且产率高、后处理简单。
下面结合具体实施方式对本发明作进一步的详细说明。
附图说明
图1为实施例1中所获化合物的核磁共振氢谱。
图2为实施例1中所获化合物的偏光织构图。
图3为实施例1中所获化合物的dsc图。
图4为实施例1中所获化合物在四氢呋喃溶液中和薄膜时的荧光发射图。
图5为实施例2中所获化合物的核磁共振氢谱。
图6为实施例2中所获化合物的偏光织构图。
图7实施例2中所获化合物的dsc图。
图8为实施例2中所获化合物在四氢呋喃溶液中和薄膜时的荧光发射图。
图9为实施例3中所获化合物的核磁共振氢谱。
图10为实施例3中所获化合物的偏光织构图。
图11为实施例3中所获化合物的dsc图。
图12为实施例3中所获化合物在四氢呋喃溶液中和薄膜时的荧光发射图。
图13为实施例4中所获化合物的核磁共振氢谱。
图14为实施例4中所获化合物的偏光织构图。
图15为实施例4中所获化合物的dsc图。
图16为实施例4中所获化合物在四氢呋喃溶液中和薄膜时的荧光发射图。
图17为实施例5中所获化合物的核磁共振氢谱。
图18为实施例5中所获化合物的偏光织构图。
图19为实施例5中所获化合物的dsc图。
图20为实施例5中所获化合物在四氢呋喃溶液中和薄膜时的荧光发射图。
图21为实施例6中所获化合物的核磁共振氢谱。
图22为实施例6中所获化合物的偏光织构图。
图23为实施例6中所获化合物的dsc图。
图24为实施例6中所获化合物在四氢呋喃溶液中和薄膜时的荧光发射图。
具体实施方式
实施例1
本发明的一种具体实施方式是:一种含氮盘状离子液晶化合物,具有下述通式i的结构,其中r1=c6h13,r2=c6h13,x为有机阴离子双三氟甲烷磺酰亚胺离子。
通式i中r1=c6h13,r2=c6h13,x为有机阴离子双三氟甲烷磺酰亚胺离子的含氮盘状离子液晶化合物的制备反应式如下:
反应式中thf为四氢呋喃;toluene为甲苯;tf2nli为双三氟甲烷磺酰亚胺锂。
3,4-二己基苯基硼酸的制备为现有技术,参见文献:chun-xialiu,huwang,jun-qidu,ke-qingzhao,pinghu,bi-qinwang,hirosatomonobe,benoîtheinrichandbertranddonnio.moleculardesignofbenzothienobenzothiophene-coredcolumnarmesogens:facilesynthesis,mesomorphism,andchargecarriermobility.j.mater.chem.c.,2018,6,4471—4478.
具体的制备步骤如下:
a、suzuki偶联反应
将1摩尔份的3,4-二溴吡啶、2.5摩尔份3,4-二己基苯基硼酸、10摩尔份的碳酸钾、0.25摩尔份的四(三苯基膦)合鈀加入到thf和h2o的混合溶液中,在氩气保护下,70℃搅拌反应24小时,反应结束后用二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙酸乙酯的体积比=10:1),得到粘稠状化合物,产率69%;
b、分子内芳烃氧化偶联反应
将a步得到的1摩尔份产物溶解在二氯甲烷中,缓慢加入溶解在硝基甲烷中的3摩尔份的三氯化铁溶液,滴加完后,室温搅拌反应3小时,反应过程中用薄层色谱跟踪至反应物反应完全,然后加入甲醇淬灭反应,用饱和食盐水洗涤,调节ph至碱性,二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙酸乙酯的体积比=10:1),石油醚重结晶,得到的白色固体化合物即为含氮苯并菲,产率65%;
c、烷基化反应
将1摩尔份的含氮苯并菲溶于甲苯中,加入5摩尔份的1-溴己烷,80℃搅拌反应24小时,反应结束后,蒸去甲苯,将剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙醇的体积比=10:1),用无水乙醇/无水甲醇重结晶,过滤,真空干燥,得到黄色固体化合物,产率83%;
d、离子交换反应(复分解反应)
将c步得到的1摩尔份的烷基化产物溶解在二氯甲烷中,加入3摩尔份的双三氟甲烷磺酰亚胺锂,加入少量蒸馏水,室温下搅拌反应2小时,反应结束后,用蒸馏水洗涤有机相3次,无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙醇的体积比=10:1),乙酸乙酯/无水乙醇重结晶,过滤,真空干燥,得到黄色固体化合物,产率85%。
以下的数据表明,采用以上本例方法制得的产物确是通式i中r1=c6h13,r2=c6h13,x为有机阴离子双三氟甲烷磺酰亚胺离子的含氮盘状离子液晶化合物。
核磁氢谱1hnmr(cdcl3,tms,600mhz)δ:9.57(s,1h,arh),8.53(d,j=4.8hz,1h,arh),8.37(d,j=4.8hz,1h,arh),7.83(s,1h,arh),7.56(s,1h,arh),7.54(s,1h,arh),7.51(s,1h,arh),4.69(s,2h,nch2),4.29(s,2h,aroch2),4.24(t,j=6.0hz,4h,aroch2),4.06(t,j=6.6hz,2h,aroch2),1.90-2.01(m,10h,ch2),1.53-1.61(m,10h,ch2),1.30-1.43(m,20h,ch2),0.93-0.97(m,12h,ch3),0.86(t,j=7.2hz,3h,ch3).
元素分析elementalanalysis:calculatedforc49h72f6n2o8s2,c59.14%,h7.29%,n2.81%;foundc59.27%,h7.36%,n2.63%.
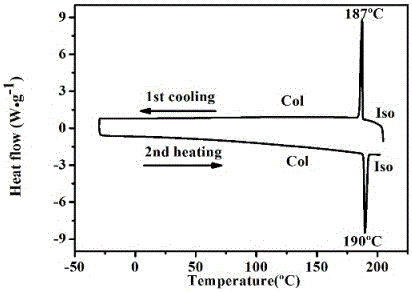
该实施例中化合物通过差示扫描量热法、偏光显微镜摄像,及小角度x-射线衍射,发现其具有典型的液晶织构和很宽的液晶相变温度范围(-30~190oc),测试了薄膜和液体的荧光发射,在溶液中和薄膜时都发黄光。
实施例2
通式i中r1=c6h13,r2=c6h13,x为有机阴离子十二烷基磺酸根离子的含氮盘状离子液晶化合物的制备反应式如下:
具体的制备步骤如下:
a、suzuki偶联反应
将1摩尔份的3,4-二溴吡啶、2.5摩尔份3,4-二己基苯基硼酸、10摩尔份的碳酸钾、0.25摩尔份的四(三苯基膦)合鈀加入到thf和h2o的混合溶液中,在氩气保护下,70℃搅拌反应36小时,反应结束后用二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙酸乙酯的体积比=10:1),得到粘稠状化合物,产率72%;
b、分子内芳烃氧化偶联反应
将a步得到的1摩尔份产物溶解在二氯甲烷中,缓慢加入溶解在硝基甲烷中的3摩尔份的三氯化铁溶液,滴加完后,室温搅拌反应3小时,反应过程中用薄层色谱跟踪至反应物反应完全,然后加入甲醇淬灭反应,用饱和食盐水洗涤,调节ph至碱性,二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙酸乙酯的体积比=10:1),石油醚重结晶,得到的白色固体化合物即为含氮苯并菲,产率66%;
c、烷基化反应
将1摩尔份的含氮苯并菲溶于甲苯中,加入3摩尔份的1-溴己烷,80℃搅拌反应36小时,反应结束后,蒸去甲苯,将剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙醇的体积比=10:1),用无水乙醇/无水甲醇重结晶,过滤,真空干燥,得到黄色固体化合物,产率80%;
d、离子交换反应(复分解反应)
将c步得到的1摩尔份的烷基化产物溶解在二氯甲烷中,加入5摩尔份的十二烷基磺酸钠,加入少量蒸馏水,室温下搅拌反应3小时,反应结束后,用蒸馏水洗涤有机相3次,无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙醇的体积比=10:1),乙酸乙酯/无水乙醇重结晶,过滤,真空干燥,得到黄色固体化合物,产率67%。
以下的数据表明,采用以上本例方法制得的产物确是通式i中r1=c6h13,r2=c6h13,x为有机阴离子十二烷基磺酸根离子的含氮盘状离子液晶化合物。
核磁氢谱1hnmr(cdcl3,tms,600mhz)δ:10.51(s,1h,arh),8.66(d,j=6.0hz,1h,arh),8.59(d,j=6.6hz,1h,arh),8.34(s,1h,arh),7.51(s,1h,arh),7.44(s,1h,arh),7.43(s,1h,arh),5.07(s,2h,nch2),4.49(t,j=5.4hz,2h,aroch2),4.22-4.25(m,4h,aroch2),4.06(t,j=6.0hz,2h,och2),2.98(t,j=7.8hz,2h,och2),1.91-2.00(m,12h,ch2),1.74-1.82(m,6h,ch2),1.53-1.66(m,8h,ch2),1.41-1.43(m,20h,ch2),1.26-1.29(m,14h,ch2),0.93-0.98(m,12h,ch3),0.88(t,j=7.2hz,3h,ch3),0.83(t,j=7.2hz,3h,ch3).
元素分析elementalanalysis:calculatedforc59h97no7s,c73.47%,h10.14%,n1.45%;foundc73.27%,h9.74%,n1.79%.
该实施例中化合物通过差示扫描量热法、偏光显微镜摄像,及小角度x-射线衍射,发现其具有典型的液晶织构和很宽的液晶相变温度范围(-30~239oc),测试了薄膜和液体的荧光发射,在溶液中和薄膜时都发黄光。
实施例3
通式i中r1=c6h13,r2=c6h13,x为手性有机阴离子l-樟脑磺酸根离子的含氮盘状离子液晶化合物的制备反应式如下:
具体的制备步骤如下:
a、suzuki偶联反应
将1摩尔份的3,4-二溴吡啶、2.5摩尔份3,4-二己基苯基硼酸、10摩尔份的碳酸钾、0.25摩尔份的四(三苯基膦)合鈀加入到thf和h2o的混合溶液中,在氩气保护下,70℃搅拌反应24小时,反应结束后用二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙酸乙酯的体积比=10:1),得到粘稠状化合物,产率69%;
b、分子内芳烃氧化偶联反应
将a步得到的1摩尔份产物溶解在二氯甲烷中,缓慢加入溶解在硝基甲烷中的2摩尔份的三氯化铁溶液,滴加完后,室温搅拌反应3小时,反应过程中用薄层色谱跟踪至反应物反应完全,然后加入甲醇淬灭反应,用饱和食盐水洗涤,调节ph至碱性,二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙酸乙酯的体积比=10:1),石油醚重结晶,得到的白色固体化合物即为含氮苯并菲,产率67%;
c、烷基化反应
将1摩尔份的含氮苯并菲溶于甲苯中,加入3摩尔份的1-溴己烷,80℃搅拌反应36小时,反应结束后,蒸去甲苯,将剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙醇的体积比=10:1),用无水乙醇/无水甲醇重结晶,过滤,真空干燥,得到黄色固体化合物,产率80%;
d、离子交换反应(复分解反应)
将c步得到的1摩尔份的烷基化产物溶解在二氯甲烷中,加入溶于水的5摩尔份l-樟脑磺酸,5摩尔份的氢氧化钠溶液,室温下搅拌反应3小时,反应结束后用二氯甲烷萃取,蒸馏水洗涤有机相3次,无水硫酸镁干燥,过滤,浓缩,无水甲醇重结晶,真空干燥,得黄色固体化合物,产率77%。
以下的数据表明,采用以上本例方法制得的产物确是通式i中r1=c6h13,r2=c6h13,x为有机阴离子l-樟脑磺酸根离子的含氮盘状离子液晶化合物。
核磁氢谱1hnmr(cdcl3,tms,400mhz)δ:10.29(s,1h,arh),8.79(d,j=6.4hz,1h,arh),8.52(d,j=6.4hz,1h,arh),8.23(s,1h,arh),7.30(s,2h,arh),7.24(s,1h,arh),5.03(s,2h,nch2),4.50(s,2h,aroch2),4.20(t,j=5.6hz,4h,aroch2),3.96(t,j=6.0hz,2h,aroch2),3.51(d,j=14.4hz,1h,ch2),2.93-2.98(m,2h,ch2),2.34-2.41(m,1h,ch2),2.06-2.10(m,1h,ch),1.89-1.97(m,12h,ch2),1.55-1.63(m,8h,ch2),1.40-1.44(m,20h,ch2),1.22-1.25(m,4h,ch2),0.92-0.99(m,18h,ch3),0.81(t,j=6.8hz,3h,ch3).
元素分析elementalanalysis:calculatedforc57h87no8s,c72.34%,h9.27%,n1.48%;foundc72.47%,h9.32%,n1.48%.
该实施例中化合物通过差示扫描量热法、偏光显微镜摄像,及小角度x-射线衍射,发现其具有典型的液晶织构和很宽的液晶相变温度范围(-30~271oc),测试了薄膜和液体的荧光发射,在溶液中和薄膜时都发黄光。
实施例4
通式i中r1=c6h13,r2=c6h13,x为无机阴离子硝酸根离子的含氮盘状离子液晶化合物的制备反应式如下:
具体实施方式如下:
a、suzuki偶联反应
将1摩尔份的3,4-二溴吡啶、2.5摩尔份3,4-二己基苯基硼酸、10摩尔份的碳酸钾、0.25摩尔份的四(三苯基膦)合鈀加入到thf和h2o的混合溶液中,在氩气保护下,70℃搅拌反应24小时,反应结束后用二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙酸乙酯的体积比=10:1),得到粘稠状化合物,产率68%;
b、分子内芳烃氧化偶联反应
将a步得到的1摩尔份产物溶解在二氯甲烷中,缓慢加入溶解在硝基甲烷中的3摩尔份的三氯化铁溶液,滴加完后,室温搅拌反应2小时,反应过程中用薄层色谱跟踪至反应物反应完全,然后加入甲醇淬灭反应,用饱和食盐水洗涤,调节ph至碱性,二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙酸乙酯的体积比=10:1),石油醚重结晶,得到的白色固体化合物即为含氮苯并菲,产率64%;
c、烷基化反应
将1摩尔份的含氮苯并菲溶于甲苯中,加入4摩尔份的1-溴己烷,80℃搅拌反应24小时,反应结束后,蒸去甲苯,将剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙醇的体积比=10:1),用无水乙醇/无水甲醇重结晶,过滤,真空干燥,得到黄色固体化合物,产率81%;
d、离子交换反应(复分解反应)
将c步得到的1摩尔份的烷基化产物溶解在二氯甲烷中,加入3摩尔份的无机盐硝酸银,加入少量蒸馏水,室温下搅拌反应3小时,反应结束后,过滤生成的溴化银沉淀,用蒸馏水洗涤有机相3次,无水硫酸镁干燥,过滤,浓缩,无水乙醇/无水甲醇重结晶,过滤,真空干燥,得到黄色固体化合物,产率81%。
以下的数据表明,采用以上本例方法制得的产物确是通式i中r1=c6h13,r2=c6h13,x为无机阴离子硝酸根离子的含氮盘状离子液晶化合物。
核磁氢谱1hnmr(cdcl3,tms,400mhz),δ:10.24(s,1h,arh),8.32(s,1h,arh),8.29(s,1h,arh),8.02(s,1h,arh),7.42(s,3h,arh),4.71(s,2h,nch2),4.31(s,2h,aroch2),4.20-4.26(m,4h,aroch2),4.08(t,j=6.0hz,2h,aroch2),1.92-1.98(m,10h,ch2),1.60-1.62(m,8h,ch2),1.40-1.44(m,18h,ch2),1.26-1.28(m,4h,ch2),0.92-0.98(m,12h,ch3),0.84(t,j=6.8hz,3h,ch3).
元素分析elementalanalysis:calculatedforc47h72n2o7,c72.64%,h9.34%,n3.60%;foundc72.17%,h9.35%,n3.54%.
该实施例中化合物通过差示扫描量热法、偏光显微镜摄像,及小角度x-射线衍射,发现其具有典型的液晶织构和很宽的液晶相变温度范围(-30~100oc),测试了薄膜和液体的荧光发射,在溶液中和薄膜时都发黄光。
实施例5
通式i中r1=c6h13,r2=ch3,x为有机阴离子双三氟甲烷磺酰亚胺离子的含氮盘状离子液晶化合物的制备反应式如下:
具体的制备步骤如下:
a、suzuki偶联反应
将1摩尔份的3,4-二溴吡啶、2.5摩尔份3,4-二己基苯基硼酸、10摩尔份的碳酸钾、0.25摩尔份的四(三苯基膦)合鈀加入到thf和h2o的混合溶液中,在氩气保护下,70℃搅拌反应24小时,反应结束后用二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙酸乙酯的体积比=10:1),得到粘稠状化合物,产率69%;
b、分子内芳烃氧化偶联反应
将a步得到的1摩尔份产物溶解在二氯甲烷中,缓慢加入溶解在硝基甲烷中的3摩尔份的三氯化铁溶液,滴加完后,室温搅拌反应3小时,反应过程中用薄层色谱跟踪至反应物反应完全,然后加入甲醇淬灭反应,用饱和食盐水洗涤,调节ph至碱性,二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙酸乙酯的体积比=10:1),石油醚重结晶,得到的白色固体化合物即为含氮苯并菲,产率65%;
c、烷基化反应
将1摩尔份的含氮苯并菲溶于甲苯中,加入5摩尔份的碘甲烷,40℃搅拌反应36小时,反应结束后,蒸去甲苯,将剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙醇的体积比=10:1),用无水乙醇/乙酸乙酯重结晶,过滤,真空干燥,得到黄色固体化合物,产率81%;
d、离子交换反应(复分解反应)
将c步得到的1摩尔份的烷基化产物溶解在二氯甲烷中,加入3摩尔份的双三氟甲烷磺酰亚胺锂,加入少量蒸馏水,室温下搅拌反应2小时,反应结束后,用蒸馏水洗涤有机相3次,无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙醇的体积比=10:1),乙酸乙酯/无水乙醇重结晶,过滤,真空干燥,得到黄色固体化合物,产率75%。
以下的数据表明,采用以上本例方法制得的产物确是通式i中r1=c6h13,r2=ch3,x为有机阴离子双三氟甲烷磺酰亚胺离子的含氮盘状离子液晶化合物。
核磁氢谱1hnmr(cdcl3,tms,400mhz)δ:8.92(s,1h,arh),8.00(s,1h,arh),7.77(s,1h,arh),7.15-7.21(m,3h,arh),7.03(s,1h,arh),4.06-4.15(m,9h),3.87(s,2h,aroch2),1.86-1.92(m,8h,ch2),1.56-1.63(m,8h,ch2),1.40-1.42(m,16h,ch2),0.94-0.97(m,12h,ch3).
元素分析elementalanalysis:calculatedforc44h62f6n2o8s2,c57.13%,h6.76%,n3.03%;foundc57.41%,h6.83%,n2.93%.
该实施例中化合物通过差示扫描量热法、偏光显微镜摄像,及小角度x-射线衍射,发现其具有典型的液晶织构和很宽的液晶相变温度范围(-30~232oc),测试了薄膜和液体的荧光发射,在溶液中和薄膜时都发黄光。
实施例6
通式i中r1=c6h13,r2=ch3,x为有机阴离子十二烷基硫酸根离子的含氮盘状离子液晶化合物的制备反应式如下:
具体的制备步骤如下:
a、suzuki偶联反应
将1摩尔份的3,4-二溴吡啶、2.5摩尔份3,4-二己基苯基硼酸、10摩尔份的碳酸钾、0.25摩尔份的四(三苯基膦)合鈀加入到thf和h2o的混合溶液中,在氩气保护下,70℃搅拌反应36小时,反应结束后用二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙酸乙酯的体积比=10:1),得到粘稠状化合物,产率70%;
b、分子内芳烃氧化偶联反应
将a步得到的1摩尔份产物溶解在二氯甲烷中,缓慢加入溶解在硝基甲烷中的3摩尔份的三氯化铁溶液,滴加完后,室温搅拌反应3小时,反应过程中用薄层色谱跟踪至反应物反应完全,然后加入甲醇淬灭反应,用饱和食盐水洗涤,调节ph至碱性,二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙酸乙酯的体积比=10:1),石油醚重结晶,得到的白色固体化合物即为含氮苯并菲,产率65%;
c、烷基化反应
将1摩尔份的含氮苯并菲溶于甲苯中,加入3摩尔份的碘甲烷,40℃搅拌反应24小时,反应结束后,蒸去甲苯,将剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙醇的体积比=10:1),用无水乙醇/乙酸乙酯重结晶,过滤,真空干燥,得到黄色固体化合物,产率78%;
d、离子交换反应(复分解反应)
将c步得到的1摩尔份的烷基化产物溶解在二氯甲烷中,加入5摩尔份的十二烷基硫酸钠,加入少量蒸馏水,室温下搅拌反应3小时,反应结束后,用蒸馏水洗涤有机相3次,无水硫酸镁干燥,过滤,浓缩,浓缩后的剩余物用硅胶柱层析分离提纯(流动相:二氯甲烷:乙醇的体积比=10:1),乙酸乙酯/无水乙醇重结晶,过滤,真空干燥,得到黄色固体化合物,产率90%。
以下的数据表明,采用以上本例方法制得的产物确是通式i中r1=c6h13,r2=ch3,x为有机阴离子十二烷基硫酸根离子的含氮盘状离子液晶化合物。
核磁氢谱1hnmr(cdcl3,tms,600mhz)δ:10.16(s,1h,arh),8.54(d,j=5.4hz,1h,arh),8.37(d,j=5.4hz,1h,arh),8.08(s,1h,arh),7.30(s,1h,arh),7.29(s,1h,arh),7.27(s,1h,arh),4.61(s,3h,ch3),4.42(t,j=5.4hz,2h,aroch2),4.15-4.22(m,6h,aroch2),3.99(t,j=6.0hz,2h,och2),1.92-2.00(m,8h,ch2),1.74-1.76(m,2h,ch2),1.61-1.64(m,8h,ch2),1.40-1.44(m,18h,ch2),1.25-1.33(m,16h,ch2),0.93-0.98(m,12h,ch3),0.87(t,j=6.6hz,3h,ch3).
元素分析elementalanalysis:calculatedforc54h87no8s,c71.25%,h9.63%,n1.54%;foundc70.62%,h9.50%,n1.53%.
该实施例中化合物通过差示扫描量热法、偏光显微镜摄像,及小角度x-射线衍射,发现其具有典型的液晶织构和很宽的液晶相变温度范围(-30~250oc),测试了薄膜和液体的荧光发射,在溶液中和薄膜时都发黄光。
本发明的盘状离子液晶化合物采用偏光显微镜摄像,差示扫描量热法,小角度x-射线衍射,发现其具有典型的液晶织构-六方柱状相织构。
本发明的盘状离子液晶化合物系列具有互变液晶相,其液晶性能结果见下表:
表中:m,m1-介晶相,col-柱状相,iso-液体。
- 还没有人留言评论。精彩留言会获得点赞!