甜叶菊衍生的分子,获得此类分子的方法,及其用途与流程
甜叶菊衍生的分子,获得此类分子的方法,及其用途
背景技术:
[0001]
由于认识到许多疾病与食用高糖食品和饮料有关,因此糖替代品正受到越来越多的关注。然而,由于尚未有定论的安全性问题,许多人造甜味剂(如甘素(dulcin)、环氨酸钠(sodiumcyclamate)和糖精)在一些国家中受到限制。因此,天然来源的无热量甜味剂变得越来越流行。甜味草药甜叶菊(stevia rebaudiana)产生大量的二萜糖苷,其特征在于高强度甜味和优于许多其它高效甜味剂那些的感官特性。
[0002]
甜叶菊(stevia rebaudiana)是属于astracea科的植物,原产于南美,且现已在世界许多地方种植(gardana等,2003;koyama等,2003;carakostas等,2008)。甜叶菊的叶天然是甜的,且在南美用于食品增甜已有数百年了(soejarto等,1982)。在日本和其它东南亚国家中甜叶菊提取物已经在商业上用于增甜食品多年(koyama等,2003)。作为天然产物,甜叶菊植物的叶子含有不同的甜味成分,称为甜菊醇糖苷。据报道,已经鉴定出甜叶菊叶提取物中通常存在40多种甜菊醇糖苷(ceunen和geuns,2013;purkayastha等,2016)。这些甜菊醇糖苷中的每一种都有其独特的味道和甜度,其可以比糖甜高达350倍,但都具有相似的分子结构,其中不同的糖基团连接糖苷配基甜菊醇(一种ent-贝壳杉烯类二萜,ent-kaurene-type diterpene)。
[0003]
甜叶菊植物的叶子含有一种混合物,该混合物含有含量范围为总干重约10%至约20%的二萜糖苷。这些二萜糖苷比糖甜约30至450倍。在结构上,许多二萜糖苷的特征在于单碱基、甜菊醇,并且不同之处在于c13和c19位存在碳水化合物残基。通常,基于干重,甜叶菊叶中发现的四种主要的甜菊醇糖苷是杜克苷a(0.3%)、莱鲍迪苷c
[0004]
(0.6-1.0%)、莱鲍迪苷a(3.8%)和甜菊糖苷(9.1%)。甜叶菊提取物中鉴定出的其它糖苷包括莱鲍迪苷b、d、e和f,甜菊双糖苷和甜茶苷(rubusoside)。
[0005]
莱鲍迪苷a和甜菊糖苷已经引起了最大的商业兴趣,并且就其作为商业高强度甜味剂的适用性进行了广泛的研究和表征。碳酸饮料中的稳定性研究证实了它们的热稳定性和ph稳定性(chang s.s.,cook,j.m.(1983)stability studies of stevioside and rebaudioside a in carbonated beverages.j.agric.food chem.31:409-412.)
[0006]
甜菊醇糖苷之间的区别不仅在于分子结构,还在于它们的味道特性。通常发现甜菊糖苷比蔗糖甜110-270倍,而莱鲍迪苷a比蔗糖甜150-320倍。莱鲍迪苷a的涩味最少、苦味最少且回味最不持久,因此在主要甜菊醇糖苷中拥有最有利的感官属性(tanaka o.(1987)improvement of taste of natural sweeteners.pure appl.chem.69:675-683;phillips k.c.(1989)stevia:steps in developing a new sweetener.in:grenby t.h.编辑,developments in sweeteners,vol.3.elsevier applied science,london.1-43.)。
[0007]
到21世纪初,只表征了有限数量的甜叶菊中甜菊醇糖苷的化学结构,包括甜菊糖苷、莱鲍迪苷a-f、杜克苷a和甜菊双糖苷(ceunen和geuns,2013)。近年来,已经从甜叶菊的叶子中报道了具有不同化学结构的许多次要甜菊醇糖苷(chaturvedula等,2011a,b,c;chaturvedula和prakash,2011a,b)。这些不同的甜菊醇糖苷,即ent-贝壳杉烯类二萜(ent-kaurene-type diterpene),通过1,2-;1,3-;1,4-或1,6-α或β-糖苷键在c-13和c-19位连接
各种糖,如葡萄糖、鼠李糖、木糖、果糖和脱氧葡萄糖(purkayastha等,2016)。
[0008]
迄今为止,甜菊糖苷的使用受到某些不良味道特性的限制,包括甘草味、苦味、涩味、甜味回味、苦味回味、甘草回味,并且随着浓度的增加而变得更加突出。这些不良的口味属性在碳酸饮料中尤为突出,在碳酸饮料中,糖的完全替代需要浓度超过600mg/l的甜菊醇糖苷。如此高浓度的甜菊醇糖苷的使用导致最终产品味道的明显恶化。
[0009]
因此,仍然需要研发天然的减热或无热量甜味剂,其提供与蔗糖的时间和风味特征相似的时间和风味特征。
[0010]
进一步仍然需要从甜叶菊植物纯化糖苷的方法。
技术实现要素:
[0011]
本发明总体上涉及新型二萜糖苷以及包含所述新型二萜糖苷的组合物和食品,以及用于纯化所述新型二萜糖苷的方法、用于制备包含所述新型二萜糖苷的组合物和食品的方法和使用新型二萜糖苷增强或改变食品的风味或甜味的方法。本发明的新型二萜糖苷从甜叶菊植物分离。
[0012]
本发明涉及甜叶菊衍生的分子、用于获得此类分子的方法以及此类分子的用途。这些甜叶菊衍生的分子可以具有或可以不具有甜菊醇主链结构,但具有与甜菊醇糖苷有些相似或基本上相似的结构。在一些情况中,这些分子具有非常不同于甜菊醇糖苷的结构。这些甜叶菊衍生的分子具有理想的味道和风味特性,这可以包括影响甜味的特性、改变风味的特性、这些特性的组合,以及其它特性。
附图说明
[0013]
图1显示了使用梯度km7的甜叶菊提取物a95的代表性分析色谱。上图和中图是ms tic(-)(质谱仪总离子流)色谱,而下图是elsd(蒸发光散射检测仪)色谱。
[0014]
图2是用于分离表1中所列的不同化合物的示意性步骤的图。
[0015]
图3是用于分离表1中所列的不同化合物的示意性步骤的图。
[0016]
图4显示了rsg1(相关的甜菊醇糖苷1)的结构。
[0017]
图5显示了rsg2(相关的甜菊醇糖苷2)的结构。
[0018]
图6显示了rsg3(相关的甜菊醇糖苷3)的结构。
[0019]
图7显示了rsg4(相关的甜菊醇糖苷4)的结构。
[0020]
图8显示了rsg5(相关的甜菊醇糖苷5)的结构。
[0021]
图9显示了rsg6(相关的甜菊醇糖苷6)的结构。
[0022]
图10显示了莱鲍迪苷t的结构。
[0023]
图11显示了莱鲍迪苷y的结构。
[0024]
图12显示了莱鲍迪苷o2的结构。
[0025]
图13显示了莱鲍迪苷c2的结构。
[0026]
图14显示了莱鲍迪苷w的结构。
[0027]
图15显示了莱鲍迪苷w2的结构。
[0028]
图16显示了莱鲍迪苷u2的结构。
[0029]
图17a显示了甜叶菊叶提取物的选定馏分(级分)的rp-hplc分析。
[0030]
图17b显示了甜叶菊叶提取物的选定馏分的elsd和ms分析。
[0031]
图17c显示了甜叶菊叶提取物的选定馏分的1h-nmr分析。
[0032]
图17d显示了莱鲍迪苷w3的结构。
[0033]
图18显示了莱鲍迪苷v的结构。
[0034]
图19显示了莱鲍迪苷u的结构。
[0035]
图20显示了莱鲍迪苷k2的结构。
[0036]
图21显示了莱鲍迪苷v2的结构。
[0037]
图22显示了rsg7(相关的甜菊醇糖苷7)的结构。
[0038]
图23显示了rsg8(相关的甜菊醇糖苷8)的结构。
[0039]
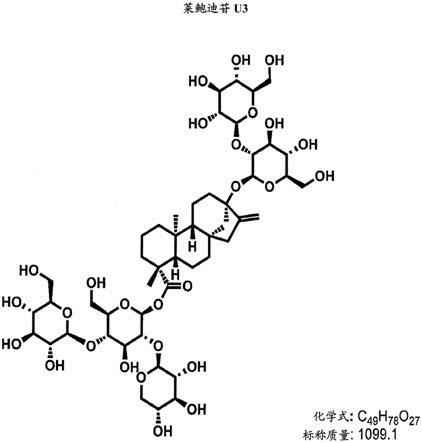
图24显示了莱鲍迪苷u3的结构
[0040]
发明详述
[0041]
本发明某些甜叶菊衍生分子的化学结构示于所附附图中。如本文所用的,“甜叶菊衍生的分子”应指获自甜叶菊种的任何品种的植物的任何部位的分子。
[0042]
这些甜叶菊衍生的分子可用于制备食品、饮料、营养食品、药物、烟草制品、化妆品、口腔卫生制品等。一些甜叶菊衍生的分子具有甜菊醇主链,并且可以被称为甜菊醇糖苷。本发明的其它甜叶菊衍生的分子具有不同的主链,但是可以具有与甜菊醇糖苷相似的性质,或者可以具有其它有益的性质。
[0043]
这些甜叶菊衍生的分子可以单独使用或与其它成分结合使用,如甜味剂、调味剂、风味改良剂等。这样的其它成分可以包括甜菊醇糖苷成分,或来自其它天然或合成来源的成分。
[0044]
获得甜叶菊衍生的分子的方法包括用于从甜叶菊液体提取甜菊醇糖苷的方法。其它方法可以包括从植物的其它部位提取,或其它提取技术和溶剂。
[0045]
以下实施例证明了本发明的某些实施方案,并且不是旨在以任何方式限制本发明的范围。
具体实施方式
[0046]
实施例1
[0047]
将可获自purecircle usa inc.of oak brook,il的标为“a95”的甜叶菊提取物用于使用以下分析方法来分离和表征主要和次要甜菊醇糖苷组分。
[0048]
1.1样品
[0049]
产品名称:甜叶菊叶提取物a95
[0050]
批号:wip a95 27a
[0051]
生产日期2016年04月02日
[0052]
1.2分析lcms(液晶质谱)
[0053]
在shimadzu单四极杆(single quad)uplc系统上进行了分析lcms(参见表1)。应用了两个不同的梯度系统(参见表2a和2b),它们头40分钟内是相同的。梯度km7用于解析所有化合物,包括已鉴定的甜叶菊糖苷#25-#29,而梯度acd1则更快,用于分析化合物#1-#24。
[0054]
通过将甜叶菊叶提取物a95(20mg)溶解于甲醇和二甲基亚砜(dmso)的1:1混合物中来制备参照样品。为了获得均匀溶液,需要进行30分钟的超声处理。将该溶液储存在4℃。
[0055]
证明了该分析系统对溶剂组成的变化非常敏感,并且在使用新一批溶剂时观察到了保留时间的变化。因此,在每个分析批次之前和之后分析参照样品,并验证保留时间的分配。
[0056]
使用梯度km7的典型分析色谱显示于图1中。
[0057]
表1:lcms系统
[0058]
hplc系统shimadzu lc-30ad,突出界面shimadzucbm-20a脱气机shimadzudgu-20a5自动加样器shimadzu sil-30ac,突出柱温箱shimadzucto-20acmsshimadzu 2020单四级杆dadshimadzu spd-m20aelsdsedere elsd-lt ii,sedex 85固定相agilent poroshell 120sb-c18 2.7μm,4.6x150mm流速0.5ml/min移动相a:水,25%乙腈,0.2%乙酸,b:乙腈
[0059]
表2:lcms梯度
[0060]
[0061][0062]
1.3重结晶
[0063]
将甜叶菊叶子提取物a95(100g,白色粉末)在65℃的温度下溶解于乙醇/水70/30中(750ml)。
[0064]
使乳白色溶液在水浴中冷却至室温,并随后通过吸滤器过滤。用乙醇洗涤收集的晶体,干燥并储存。将母液和洗涤溶液分开保存,并且在真空下除去各自的溶剂。
[0065]
1.4反相mplc(中压液相色谱)
[0066]
将各自的样品(15g)溶解于甲醇中,加入硅藻土(30g)并通过旋转蒸发仪除去溶剂。将固定的样品转移至玻璃柱中并构建表3中所述的mplc系统。基于时间的分馏(分馏分离)产生18个馏分(级分)(每个4min)。溶剂和梯度描述于表3中。
[0067]
表3:mplc-系统和梯度
[0068]
泵系统 界面模块scpa馏分收集器labomaticlabocolvario 2000plus固定相polygoprep c18,50-60μm,玻璃柱50x250mm
[0069][0070][0071]
1.5正相色谱
[0072]
将各自的样品(20g)溶解在甲醇中,加入二氧化硅(40g),并通过旋转蒸发仪除去溶剂。将固定的样品转移到玻璃柱中,并构建表4中所述的高压液相色谱(hplc)系统。通过用乙酸乙酯/甲醇1:1洗涤从转移柱中除去空气。基于时间的分馏产生90个馏分(每个0.5min),将这些馏分基于分馏过程中生成的uv和elsd数据进行合并。通过lcms分析所得的馏分。溶剂和梯度描述于表4中。
[0073]
表4:制备性hplc系统2(htp-ii,np-分馏)
[0074][0075]
1.6反相hplc
[0076]
将各自的样品(最大3.5g)溶解在甲醇中,加入c-18rp材料,并通过旋转蒸发仪除去溶剂。将固定的样品转移到色谱柱中,并构建表5中所述的hplc系统。基于时间的分馏产生120个馏分(每个27sec),将这些馏分基于分馏过程中产生的uv和elsd数据进行合并。通过lcms分析所得的馏分。溶剂和梯度描述于表5中。
[0077]
表5:制备性hplc系统3(sepbox)
[0078]
[0079]
[0080]
[0081][0082]
1.7 hilic(亲水性相互作用液相色谱)
[0083]
将各自的样品溶解于2ml的溶剂a和b的3:1混合物中(参见表6)。9.95分钟后进行样品注射。基于时间的分馏产生96个馏分(每个43sec,从18分钟后开始),基于分馏过程中生成的uv和elsd数据将这些馏分进行合并。通过lcms分析所得馏分。溶剂和梯度描述于表6中。
[0084]
表6:制备性hplc系统1(htp-i,hilic-馏分)
[0085][0086]
1.8 nmr(核磁共振)
[0087]
使用bruker 500mhz nmr光谱仪通过nmr光谱鉴定了分离的化合物。糖苷配基的鉴定基于参考1h-nmr光谱,使用c17、c18和c20质子信号作为主要指示。如化合物#4和#18所示,尤其是c20质子转移表明了改变。使用h-h-cosy、hsqc和hmbc,以及使用已知甜菊苷作为参照的文献光谱的实验阐明了糖苷。
[0088]
1.9结果
[0089]
图1显示了使用如上所述的分析方法含有表7中鉴定的主要峰的hplc图。分离表7中的不同化合物的示意步骤显示于图2和图3中。
[0090]
表7
[0091][0092]
使用实施例1的方法分离的新型甜叶菊叶衍生的分子的列表显示于表8和表9中。
[0093]
表8:相关的甜菊醇糖苷组分
[0094][0095]
表9:新型甜菊醇糖苷组分
[0096]
甜菊醇糖苷(峰id)分子量简易分子式分子式停留时间(min)莱鲍迪苷t(#6)1129svgal1g4c
50
h
80
o
28
8.95莱鲍迪苷y(#acd 2)1261sva1g5c
55
h
88
o
32-莱鲍迪苷o2(#acd 14)1437svr1g6c
62
h
100
o
37-莱鲍迪苷c2(#13)951svr1g3c
44
h
70
o
22
15.79莱鲍迪苷w(#15)1099sva1g4c
49
h
78
o
27
17.93莱鲍迪苷w2(#17b)1099sva1g4c
49
h
78
o
27
na莱鲍迪苷u2(#17a)1099svx1g4c
49
h
78
o
27
18.8莱鲍迪苷w3(#19)1099sva1g4c
49
h
78
o
27
20.26莱鲍迪苷v(#acd6)1261svx1g5c
55
h
88
o
32
20.95莱鲍迪苷u(#20)1099svx1g4c
49
h
78
o
27
21.14莱鲍迪苷k2(#21)1113svr1g4c
50
h
80
o
27
23.31莱鲍迪苷v2(#22)1261svx1g5c
55
h
88
o
32
25.51莱鲍迪苷u31099svx1g4c
49
h
78
o
27
18.80
[0097]
实施例2:新化合物的鉴定和表征
[0098]
这个实施例概述了作为实例的莱鲍迪苷w3(#19)的分离、鉴定和表征。对于所有新的甜菊醇糖苷分子进行了相似的分析。
[0099]
分离
[0100]
根据1.3节(实施例1)中描述的方法,将100g甜叶菊叶提取物a95重结晶,从母液产生33.2g富集的次要化合物。使用梯度a(参见表4),使用1.5节中所述的正相色谱将富集的次要化合物分馏。馏分49-60产生了1.32g富集的次要化合物,使用梯度l,使用根据1.4节的反相hplc,将其进一步分馏。
[0101]
rp(反相)-hplc&lcms
[0102]
馏分51+52由矩形标记(图17a),elsd迹线和uv迹线产生了37.5mg#19。使用制备性rp-hplc色谱,馏分66+67(图17b)产生了3.85g富集的次要化合物。根据3.2节通过lcms分析了馏分66+67(参见图17b)。获得了37.5mg的化合物#19,具有89%纯度(elsd)。
[0103]
nmr
[0104]
在d
4-甲醇中在500mhz bruker-nmr上,通过nmr测定了化合物#19的结构(δ
c
=48.5ppm;δ
h
=3.3ppm)。数据显示于表10中,并且nmr分析显示于图17c中。化合物#19的结构
显示于图17d中。
[0105]
表10:1h-和
13
c-nmr信号的分配(基于hh-cosy、hsqc、hmbc和hsqc-tocsy实验)
[0106]
[0107][0108]
认为以上鉴定的这些次要分子中的每一个对含有基于甜叶菊的成分的产品的甜度、味道和风味特征具有许多令人满意的效果,所述分子优选纯度水平范围为80-99%,包
括90-95%纯度,99%纯度和89%纯度或更高,作为分离的分子或结合其它甜叶菊衍生的分子组合。这些分子可用于食品、饮料、营养食品、药物和其它可食用或消费产品中赋予特定的味道或修饰风味,或两者兼具。
[0109]
应当理解,本文中所示的前述和具体实施方案仅是对本发明的最佳实施方式及其原理的说明,并且本领域技术人员可以在不脱离本发明的精神和范围的情况下容易地进行修改和增加,因此将理解本发明的保护范围仅由所附权利要求范围来限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1