一种高纯度肝素钠的制备工艺的制作方法
本发明涉及医药技术领域,具体为一种高纯度肝素钠的制备工艺。
背景技术:
肝素广泛存在于哺乳动动的内脏和肠粘膜中,主要由d-葡糖胺,l-艾杜糖醛酸,m-乙酰葡萄糖胺及d-葡糖醛酸交替组成的粘多糖硫酸酯,作为一种天然抗凝血物质,在抗凝血、促进质蛋白酶释放和补体溶细胞体系等方面具有良好的活性,广泛用于流行性脑炎、败血症、血栓塞、急性心肌梗塞、动脉硬化等治疗,肝素易溶于水,不溶于乙醇、丙酮等有机溶剂,分子结构单元中含有5个硫酸基和2个羟基,呈强酸性,为聚阴离子,能与阳离子发生反应生成盐,由于肝素在动物体中主要以肝素钠-蛋白质复合物的形式存在,故使得目前动物体中提取的粗品肝素钠中残留一定量的蛋白质,对肝素钠的活性产生一定影响。
目前肝素的精制过程主要是对粗品肝素钠进行除蛋白、核酸等物质提纯和脱色,达到国家药典标准要求,国内外发表了许多除肝素中丝氨酸的方法,但是均需分离特定分子量的肝素片段,然后除去特定肝素片段的丝氨酸残基,无法达到工业化生产要求,而现有肝素提取纯化方法大多采取酸碱处理去除杂质蛋白、核酸等物质,采用高锰酸钾氧化和过氧化氢氧化去除色素类物质,但是高锰酸钾氧化法会产生二氧化锰吸附肝素的问题,使肝素活性损失大,导致肝素钠回收率低、色泽差,过氧化氢氧化法,因产品色泽较好,目前被广泛应用,但由于现有技术对氧化剂加入量及反应条件难以控制,往往在实际生产中会加入过量的氧化剂,而反应条件也不适宜,最终对肝素钠结构的破坏,使其收率低,活性低,难以得到较高纯度的精品肝素钠产品。
技术实现要素:
(一)解决的技术问题
针对现有技术的不足,本发明提供了一种高纯度肝素钠的制备工艺,解决了现有肝素钠精制时因对肝素钠结构产生破坏而难以得到高纯度的肝素钠,易对肝素钠结构造成破坏,使其收率低,活性低的问题。
(二)技术方案
为实现以上目的,本发明通过以下技术方案予以实现:一种高纯度肝素钠的制备工艺,具体包括以下步骤:
s1、酶解盐解除蛋白:将10-12倍肝素钠粗品重量的纯化水升温至50.0-60.0℃,搅拌状态下加入纯化水体积2.0%-5.0%的肠衣盐,溶解后加入肝素钠粗品,继续搅拌溶解至少1小时,溶解完毕后,将料液温度降至35.0-45.0℃,加入溶解好的胰酶,搅拌均匀,控制温度35.0-45.0℃,用氢氧化钠溶液调节ph值至7-9,保温酶解反应3.0-5.0小时,加入料液体积0.3%的无水碳酸钠和0.25%的亚硫酸氢钠,升温至50.0-70.0℃,加入溶解好的无水氯化钙,升温至80.0-90.0℃,保温反应30-60分钟,加入无水碳酸钠,保温搅拌反应30-60分钟后过滤,用hplc法对过滤去除杂质进行检测分析;
s2、一次氧化:调节滤液温度至25.0-40.0℃,用4mol/l氢氧化钠溶液调节料液ph值至10-11,再加入料液体积0.3%的无水碳酸钠和0.25%的无水硫酸钠,按料液体积的2.0%加入氯化钠,加入料液体积2.0%的过氧化氢氧化12-18h;
s3、沉淀:氧化完毕后,过滤料液,按料液体积:乙醇体积1:1.0-1:1.2加入已升温至40.0-50.0℃的乙醇,控制料液乙醇浓度40±2%,温度为35.0-40.0℃,保温静置沉淀4±1h,抽去上清液,将上清中杂质复沉,用hplc法对复沉杂质进行检测分析;
s4、二次氧化:向氧化沉淀罐ⅱ中加入肝素钠粗品重量6.9-7.3倍的纯化水,调节料液温度至25.0-30.0℃,用4mol/l氢氧化钠溶液调节料液ph值至10-11,再加入料液体积0.3%的无水碳酸钠和0.25%的无水硫酸钠,搅拌溶解,再按料液体积的2.0%加入氯化钠,加入料液体积2.0%的过氧化氢氧化12-18h;
s5、再次沉淀:氧化完毕后,过滤料液,按料液体积:乙醇体积=1:1.1-1:1.3加入已升温至40.0-50.0℃的乙醇,控制料液乙醇浓度46±2%,温度为35.0-40.0℃,保温静置沉淀4±1h,抽去上清液,将上清中杂质复沉,用hplc法对复沉杂质进行检测分析;
s6、冷冻干燥:用肝素钠粗品3倍量的纯化水使沉淀物充分溶解,用4mol/l盐酸将ph值调到6.0±0.5,过滤,冷冻干燥,即可得到高纯度肝素钠。
优选的,所述步骤s1中胰蛋白酶的比例为粗品重量与效价比的1.2-1.8倍。
优选的,所述步骤s1中无水氯化钙浓度为2.0%-3.0%。
优选的,所述步骤s1中无水碳酸钠浓度为2.0%-3.0%。
优选的,所述步骤s2和步骤s4中过氧化氢的原浓度均为30%。
本发明提供的肝素钠的制备方法,融合酶解氧化为一体,在粗品肝素钠中加入蛋白酶将其中含有的蛋白进行酶解,过滤去除蛋白,再通过一次氧化沉淀,去除硫酸皮肤素和硫酸软骨素,通过二次氧化沉淀去除硫酸乙酰肝素,在氧化过程中,加入保护剂-无水碳酸钠和无水硫酸钠,多次氧化,避免肝素结构受到破坏,同时将结合在肝素上所有得杂质分离去除,最后对得到的肝素钠溶液,冷冻干燥,得到纯品,此方法保持了肝素结构完整性,所得肝素钠纯度高,杂质少,在此发明中,我们研究了在酶解和氧化过程中,保护剂对产品质量的影响,确定了保护剂的重要性,研究实例如下:
研究实例1:将10倍肝素钠粗品重量的纯化水升温至50.0℃,搅拌状态下加入纯化水体积2.0%的肠衣盐,溶解后加入肝素钠粗品,继续搅拌溶解1小时,降温至35.0℃,加入溶解好的胰酶,搅拌均匀,控制温度35.0℃,滴加4mol/l的氢氧化钠溶液,调节ph值至7,保温酶解反应3.0小时;升温至50.0℃,加入溶解好的无水氯化钙,升温至80.0℃,保温反应30分钟,加入溶液体积2.0%无水碳酸钠,保温搅拌反应30分钟后过滤,沉淀,沉淀物脱水干燥,记样品1。
研究实例2:将10倍肝素钠粗品重量的纯化水升温至50.0℃,搅拌状态下加入纯化水体积2.0%的肠衣盐,溶解后加入肝素钠粗品,继续搅拌溶解1小时,降温至35.0℃,加入溶解好的胰酶,搅拌均匀,控制温度35.0℃,滴加4mol/l的氢氧化钠溶液,调节ph值至7,保温酶解反应3.0小时,加入溶液体积0.3%浓度的无水碳酸钠和0.3%亚硫酸氢钠,升温至50.0℃,加入溶解好的无水氯化钙,升温至80.0℃,保温反应30分钟,加入溶液体积2.0%无水碳酸钠,保温搅拌反应30分钟后过滤,沉淀,沉淀物脱水干燥,记样品2。
研究实例3:将10倍肝素钠粗品重量的纯化水升温至50.0℃,搅拌状态下加入纯化水体积2.0%的肠衣盐,溶解后加入肝素钠粗品,继续搅拌溶解1小时,降温至35.0℃,加入溶解好的胰酶,搅拌均匀,控制温度35.0℃,滴加4mol/l的氢氧化钠溶液,调节ph值至7,保温酶解反应3.0小时,加入溶液体积0.3%浓度的无水碳酸钠和0.25%亚硫酸氢钠,升温至50.0℃,加入溶解好的无水氯化钙,升温至80.0℃,保温反应30分钟,加入溶液体积2.0%无水碳酸钠,保温搅拌反应30分钟后过滤,沉淀,沉淀物脱水干燥,记样品3。
通过对三批样品的检测结果,发现在酶解过程中,如果不加入任何保护剂,样品活性较低,在酶解过程中破坏了肝素结构,而加入溶液体积0.3%浓度的无水碳酸钠和0.3%亚硫酸氢钠的样品2活性低于样品3,最终确定了酶解过程中保护剂的种类与用量。
研究实例4:将样品合并溶解,调节料液温度至25.0℃,用4mol/l氢氧化钠溶液调节料液ph值至10,取出三分之一料液,按料液体积的2.0%加入氯化钠,加入料液体积2.0%的过氧化氢(原浓度为30%)氧化12h,过滤,按料液体积:乙醇体积=1:1.0-1:1.2加入已升温至40.0℃的乙醇,控制料液乙醇浓度42%,温度为35.0℃,保温静置沉淀4h,沉淀脱水干燥,记样品4。
研究实例5:另三分之一料液加入料液体积0.3%的无水碳酸钠和0.3%的无水硫酸钠,按料液体积的2.0%加入氯化钠,加入料液体积2.0%的过氧化氢(原浓度为30%)氧化12h,过滤,按料液体积:乙醇体积=1:1.0-1:1.2加入已升温至40.0℃的乙醇,控制料液乙醇浓度42%,温度为35.0℃,保温静置沉淀4h,沉淀脱水干燥,记样品5。
研究实例6:剩余三分之一料液加入料液体积0.3%的无水碳酸钠和0.25%的无水硫酸钠,按料液体积的2.0%加入氯化钠,加入料液体积2.0%的过氧化氢(原浓度为30%)氧化12h,过滤,按料液体积:乙醇体积=1:1.0-1:1.2加入已升温至40.0℃的乙醇,控制料液乙醇浓度42%,温度为35.0℃,保温静置沉淀4h,沉淀脱水干燥,记样品6。
通过对三批样品的检测结果,发现在氧化过程中,如果不加入任何保护剂,样品活性较低,在氧化过程中破坏了肝素结构;而加入溶液体积0.3%浓度的无水碳酸钠和0.3%无水硫酸钠的样品5活性低于样品6,最终确定了氧化过程中保护剂的种类与用量。
名词解释:
肠衣盐:四川自贡井矿盐品种之一,用于畜产部门加工处理肠衣。
hp:肝素,由于在电泳过程中,移动速度较慢,也称为慢移动肝素(sm-h)。
ds:硫酸皮肤素。
hs:硫酸乙酰肝素,由于在电泳过程中,移动速度较快,也称为快移动肝素(fm-h)。
cs:硫酸软骨素。
(三)有益效果
本发明提供了一种高纯度肝素钠的制备工艺。与现有技术相比具备以下有益效果:该高纯度肝素钠的制备工艺,具体包括以下步骤:s1、酶解盐解除蛋白:将10-12倍肝素钠粗品重量的纯化水升温至50.0-60.0℃,搅拌状态下加入纯化水体积2.0%-5.0%的肠衣盐,溶解后加入肝素钠粗品,继续搅拌溶解至少1小时,溶解完毕后,将料液温度降至35.0-45.0℃,加入溶解好的胰酶,搅拌均匀,s2、一次氧化:调节滤液温度至25.0-40.0℃,用4mol/l氢氧化钠溶液调节料液ph值至10-11,再加入料液体积0.3%的无水碳酸钠和0.25%的无水硫酸钠,按料液体积的2.0%加入氯化钠,加入料液体积2.0%的过氧化氢氧化12-18h,s3、沉淀:氧化完毕后,过滤料液,按料液体积:乙醇体积1:1.0-1:1.2加入已升温至40.0-50.0℃的乙醇,控制料液乙醇浓度40±2%,温度为35.0-40.0℃,保温静置沉淀4±1h,抽去上清液,将上清中杂质复沉,用hplc法对复沉杂质进行检测分析,s4、二次氧化:向氧化沉淀罐ⅱ中加入肝素钠粗品重量6.9-7.3倍的纯化水,调节料液温度至25.0-30.0℃,用4mol/l氢氧化钠溶液调节料液ph值至10-11,再加入料液体积0.3%的无水碳酸钠和0.25%的无水硫酸钠,搅拌溶解,再按料液体积的2.0%加入氯化钠,加入料液体积2.0%的过氧化氢氧化12-18h,s5、再次沉淀:氧化完毕后,过滤料液,按料液体积:乙醇体积=1:1.1-1:1.3加入已升温至40.0-50.0℃的乙醇,控制料液乙醇浓度46±2%,温度为35.0-40.0℃,保温静置沉淀4±1h,抽去上清液,将上清中杂质复沉,用hplc法对复沉杂质进行检测分析,s6、冷冻干燥:用肝素钠粗品3倍量的纯化水使沉淀物充分溶解,用4mol/l盐酸将ph值调到6.0±0.5,过滤,冷冻干燥,即可得到高纯度肝素钠,可实现通过采取酶解盐解一体式解离方式,且在解离过程中,加入无水碳酸钠和无水亚硫酸钠并以特定的比例配制,在高温解离过程中,保护肝素不受破坏,运用不同浓度乙醇除去不同杂质,在氧化过程中,加入无水碳酸钠和无水硫酸钠,以特定比例配制,解决了现有肝素钠精制时因对肝素钠结构产生破坏而难以得到高纯度的肝素钠的问题,同时运用hplc方法,对各步骤中去除的杂质进行检测,操作简单,产品收率高,更有效的控制成品质量。
附图说明
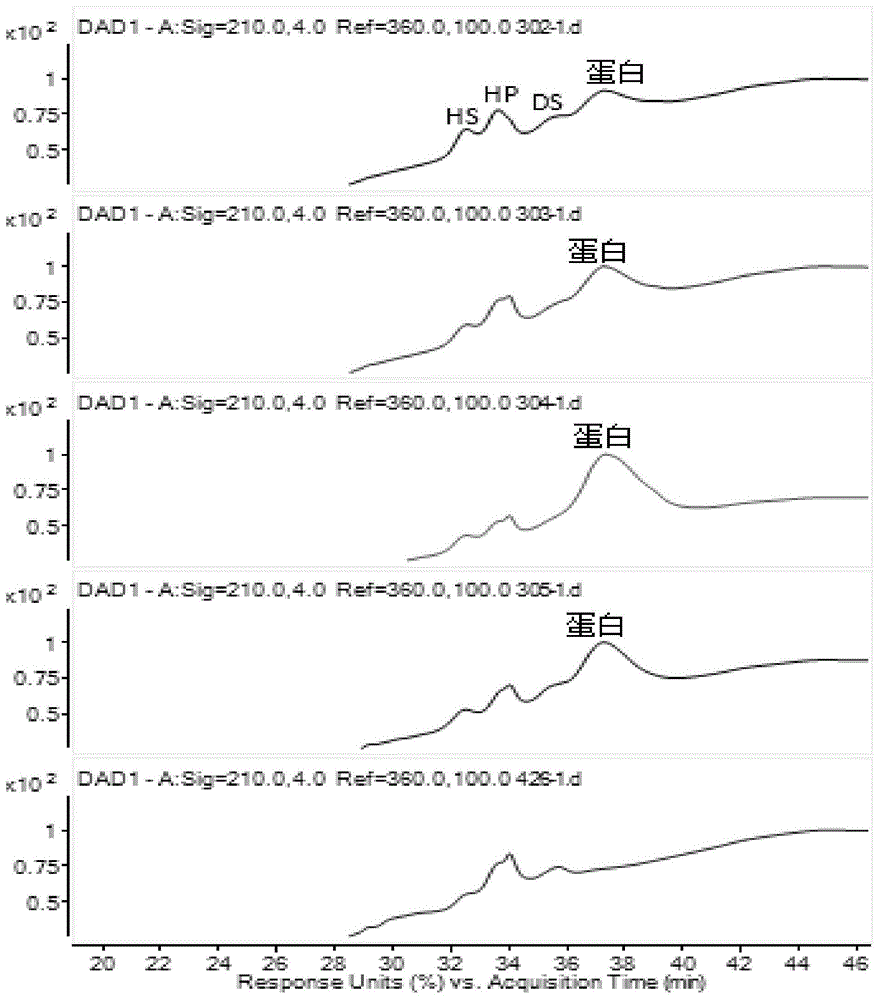
图1为本发明hplc法对各组分峰的定位示意图;
图2为本发明hplc法制备肝素钠除去杂质的分析图;
图3为本发明对比例hplc法对样品的检测峰图谱图;
图4为本发明对比例hplc法对样品的检测峰值图;
图5为本发明研究实例三的实验结果表图;
图6为本发明研究实例六的实验结果表图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
请参阅图1-5,本发明实施例提供两种技术方案:一种高纯度肝素钠的制备工艺,具体包括以下实施例:
实施例1
s1、将10倍肝素钠粗品重量的纯化水升温至50.0℃,搅拌状态下加入纯化水体积2.0%的肠衣盐,溶解后加入肝素钠粗品,继续搅拌溶解1小时,降温至35.0℃,加入肝素钠粗品重量与效价比值的1.2倍溶解好的胰酶,搅拌均匀,控制温度35.0℃,滴加4mol/l的氢氧化钠溶液,调节ph值至7,保温酶解反应3.0h,加入溶液体积0.3%浓度的无水碳酸钠和0.25%浓度的亚硫酸氢钠,升温至50.0℃,加入溶液体积2.0%无水氯化钙,升温至80.0℃,保温反应30分钟,冷却水降温过滤,加入溶液体积2.0%无水碳酸钠,保温搅拌反应30min后过滤,同时运用hplc分析方法,对过滤的杂质进行分析,记meifud。
s2、将步骤s1得到的溶液调温至25.0℃,用4mol/l氢氧化钠溶液调节料液ph值至10,再加入料液体积0.3%的无水碳酸钠和0.25%的无水硫酸钠,按料液体积的2.0%加入氯化钠,加入料液体积2.0%的过氧化氢(原浓度为30%)氧化12h。
s3、将步骤s2得到的溶液过滤,按料液体积:乙醇体积=1:1.0加入已升温至40.0℃的乙醇,控制料液酒精度38%,温度为35.0℃,保温静置沉淀3.0h,同时将上清液复沉,复沉物即为去除的杂质,运用hplc分析方法,对杂质进行检测分析,记1fu.d。
s4、将步骤s3得到的沉淀用6.9倍的纯化水溶解,调节料液温度至25.0℃,用4mol/l氢氧化钠溶液调节料液ph值至11,再加入料液体积0.3%的无水碳酸钠和0.25%的无水硫酸钠,搅拌溶解,再按料液体积的2.0%加入氯化钠,加入料液体积2.0%的过氧化氢(原浓度为30%)氧化12h。
s5、将步骤4得到的溶液过滤,按料液体积:乙醇体积=1:1.1加入已升温至40.0℃的乙醇,控制料液酒精度44%,温度为35.0℃,保温静置沉淀3.0h,同时将上清液复沉,复沉物即为去除的杂质,运用新的分析方法,对杂质进行检测分析,记2.3fu.d。
s6、冷冻干燥:将步骤7得到的沉淀用肝素钠粗品3倍量的纯化水使沉淀物充分溶解,用4mol/l盐酸将ph值调到5.5,过滤、冷冻干燥,得到肝素钠成品,记3chen.d。
实施例2
s1、将12倍肝素钠粗品重量的纯化水升温至60.0℃,搅拌状态下加入纯化水体积5.0%的肠衣盐,溶解后加入肝素钠粗品,继续搅拌溶解1.5小时,降温至45.0℃,加入肝素钠粗品重量与效价比值的1.8倍溶解好的胰酶,搅拌均匀,控制温度45.0℃,滴加4mol/l的氢氧化钠溶液,调节ph值至9,保温酶解反应5h,再加入料液体积0.3%的无水碳酸钠和0.25%的亚硫酸氢钠,升温至70.0℃,加入溶液体积3.0%无水氯化钙,升温至90.0℃,保温反应60分钟,加入溶液体积3.0%无水碳酸钠,保温搅拌反应60min后过滤。
s2、将步骤1得到的溶液,调节温度至40.0℃,用4mol/l氢氧化钠溶液调节料液ph值至11,再加入料液体积0.3%的无水碳酸钠和0.25%的无水硫酸钠,按料液体积的2.0%加入氯化钠,加入料液体积2.0%的过氧化氢(过氧化氢浓度为30%)氧化18h。
s3、将步骤2得到的溶液过滤,按料液体积:乙醇体积=1:1.2加入已升温至50.0℃的乙醇,控制料液乙醇浓度42%,温度为40.0℃,保温静置沉淀5h。
s4、将步骤3得到的沉淀用7.3倍重量的纯化水溶解,调节料液温度至30.0℃,用4mol/l氢氧化钠溶液调节料液ph值至11,再加入料液体积0.3%的无水碳酸钠和0.25%的无水硫酸钠搅拌溶解,再按料液体积的2.0%加入氯化钠,加入料液体积2.0%的过氧化氢(过氧化氢浓度为30%)氧化18h。
s5、将步骤4得到的溶液过滤,按料液体积:乙醇体积=1:1.3加入已升温至50.0℃的乙醇,控制料液乙醇浓度48%,温度为40.0℃,保温静置沉淀5h。
s6、冷冻干燥:将步骤5得到的沉淀用3倍量的纯化水充分溶解,用4mol/l盐酸将ph值调到6.5,过滤、冷冻干燥,得到肝素钠成品,记dongganchen.d。
上述所述,即为本发明的实施例,与其他发明方法样品进行对比,本发明制备的肝素钠,不含其他杂志,样品纯度最高,附图2是通过检测图谱与定位图谱对比分析,酶解盐解工序除去的杂质记-meifu.d为cs,一次氧化沉淀,通过酒精度控制,除去的杂质记-1fu.d,为大量的ds,少量的hs、cs;二次氧化沉淀通过酒精度控制,除去的杂质记23fu.d,为大量的hs和少量的ds。
对比例:专利(cn201810189164)发明实施例
1)溶解:将肝素钠粗品溶于3.5%的氯化钠溶液中,并加热至50℃,用naoh溶液调节ph至8.2,静置2.5h后抽滤,除不溶性杂质,得澄清的滤液;
2)酶解:在步骤1)中所述的澄清的滤液中加入蛋白酶,调节ph至8,将温度控制在42℃,酶解时间5.5h;
3)调酸:加入亚硫酸氢钠作为保护剂,加入盐酸调节ph至1.8,维持温度在21℃,离心脱除蛋白;
4)氧化:在步骤3)所述的离心脱除蛋白的滤液中加入过氧化氢,过氧化氢的加入量为滤液质量的2.5%,调节ph至8.8,将温度控制在26℃,进行氧化反应14.5h;
5)沉淀:将步骤4)所述的氧化反应完成的液体中加入盐酸调节ph至6.5,加入与液体等量的95%的乙醇进行沉淀,沉淀时间为12.5h,抽出上清液,得到沉淀物,将沉淀物慢慢加入到丙酮中脱水二次;
6)干燥:将步骤5)所述的脱水后的沉淀物在58℃下真空烘干5h,得到精致肝素钠记2chen.d。
更进一步地,步骤1)中肝素钠粗品与氯化钠的比例为1:14。
更进一步地,步骤2)中所述的蛋白酶的加入量为滤液质量的0.08%。
本实例即为专利(cn201810189164)下的制备方法,所制备的样品颜色偏黄,且用到丙酮,不利于大生产,且对其样品进行检测,含有其他杂质,样品纯度不高,如附图3和附图4所述。
通过对本发明与专利(cn201810189164)的样品质量对比,可以清楚的发现,样品2chen.d即专利(cn201810189164)的样品有很小的杂质峰,其杂质为硫酸乙酰肝素,而样品3chen.d和dongganchen.d即本发明两个实例制备的样品,只有一个肝素峰,不含其他杂质,主要是因为本发明在酶解过程中,以特定比例的无水碳酸钠和亚硫酸氢钠作为保护剂,不仅使高温下保持了稳定的碱性特性,使得蛋白彻底变性,而且又不会破坏肝素钠结构,在氧化过程中,同样以特定比例的无水碳酸钠和无水硫酸钠作为保护剂,在氧化过程中,不仅持续的提供了氧化所需的碱性条件,使得氧化效果稳定持续的进行,氧化效果更佳,同时无水硫酸钠的加入在氧化过程中提供了大量的硫酸根离子,这样就保证了即使更加剧烈的氧化也无法破坏肝素结构,氧化更充分,其次我们通过分级沉淀,运用不同沉淀酒精度,对不同杂质进行了去除,在较低酒精浓度下,去除了硫酸皮肤素,提高酒精浓度,去除硫酸乙酰肝素,这样就保证了产品的高纯度,因此,通过本发明生产方法制备的肝素钠,不含其他杂质,样品纯度最高,附图3中2chen.d为专利(cn201810189164)下制备方法制备的样品,3chen.d和dongganchen.d为本发明两个实例制备的样品,附图4中2chen.d为专利(cn201810189164)下制备方法制备的样品,3chen.d和dongganchen.d为本发明两个实例制备的样品。
需要说明的是,在本文中,诸如第一和第二等之类的关系术语仅仅用来将一个实体或者操作与另一个实体或操作区分开来,而不一定要求或者暗示这些实体或操作之间存在任何这种实际的关系或者顺序。而且,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
- 还没有人留言评论。精彩留言会获得点赞!