一种雄甾-2-烯-17-酮的制备方法与流程
本发明属于甾体激素药物中间体的制备工艺技术,具体涉及到一种生产罗库溴铵、维库溴铵等多种常见肌肉松弛类甾体激素药物的关键中间体雄甾-2-烯-17-酮的制备方法。
背景技术:
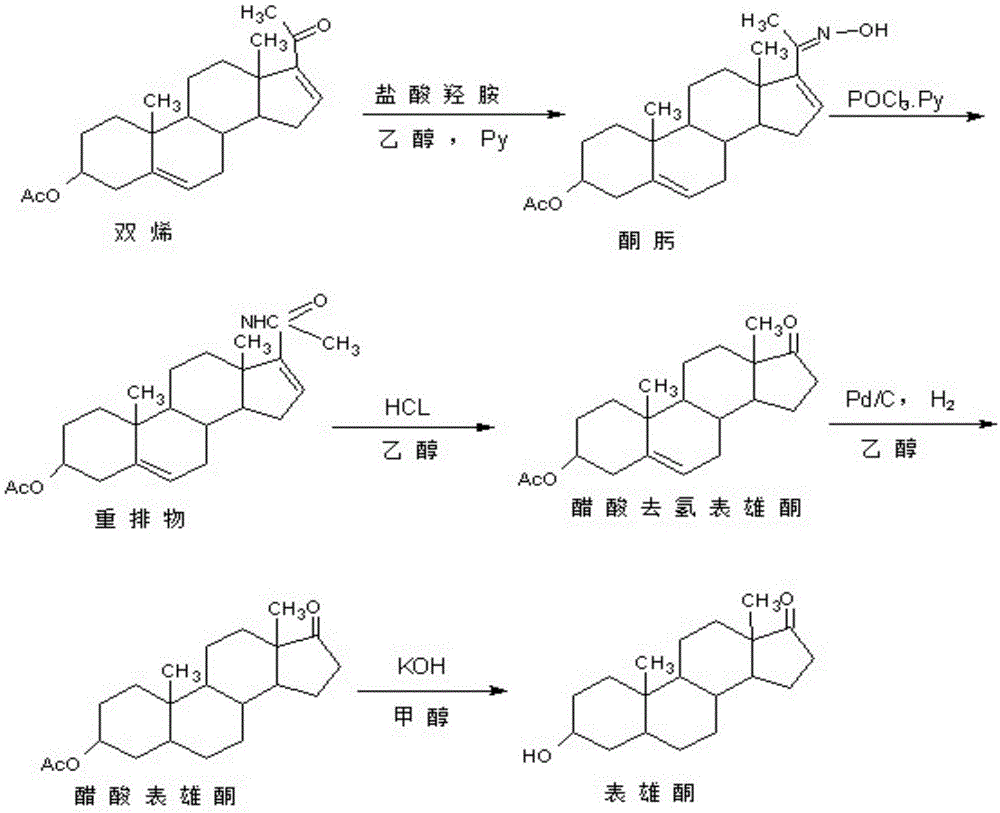
雄甾-2-烯-17-酮,别名罗库溴铵中间体lk-2,是一种生产甾体激素药物的通用中间体。以其为原料,可以生产罗库溴铵、维库溴铵等多种常见肌肉松弛类甾体激素药物。而罗库溴铵、维库溴铵等常见肌肉松弛类甾体激素药物,是一类临床上用于外科手术实施麻醉的重要辅助用药。雄甾-2-烯-17-酮经17位烯醇酯化、双环氧化、与不同的脂环胺开环、还原、双酯化、溴甲基胺化等6步反应就得到不同的溴胺类肌肉松弛类甾体激素药物。雄甾-2-烯-17-酮的传统生产方法,是以从薯蓣植物中提取的薯蓣皂素,经保护、氧化裂解、消除,获得的关键中间体醋酸妊娠双烯醇酮(简称双烯)为原料,经肟化、重排、酸水解、氢化、碱水解五步反应制得表雄酮,接着将表雄酮分子中3位羟基在碱催化下与酰氯或酸酐反应生成酯,再将其分子中形成的3位酯在碱催化下高温消除,就得到目标产物雄甾-2-烯-17-酮。其中的酯化工艺,废水较多,且不易处理;更重要的是,碱催化消除反应时,温度太低,难以发生消除反应,温度高,副反应特别大,因为消除反应有两个消除方向,分别形成目标产物雄甾-2-烯-17-酮和副产物雄甾-3-烯-17-酮,由于主副产物互为同分异构体,精制非常困难,导致雄甾-2-烯-17-酮合成收率很低,生产成本高,从而使得罗库溴铵、维库溴铵等常见肌肉松弛类甾体激素药物的生产成本与市场价格居高不下。
专利cn101058598b公开了将表雄酮溶解于苯中,加入催化剂,搅拌加热回流8~12小时,制得雄甾-2-烯-17-酮。该方法能在酸催化剂的催化下一步反应由表雄酮制备雄甾-2-烯-17-酮。但其中并未公开其原料表雄酮的制备方法。
专利cn101684139a公开了表雄酮与对甲苯磺酰氯进行酯化反应,生成表雄酮磺酰酯;与2,6-二甲基吡啶在稀硫酸作用下进行消除脱水反应生成雄甾-2-烯-17-酮。但是消除反应时的副反应大、杂质多且杂质难精制、合成收率很低。
技术实现要素:
因此,针对目前雄甾-2-烯-17-酮的现有工艺技术中消除反应副反应大,副产物量大,杂质难精制,合成收率低,生产成本高等诸多缺陷,本发明的目的就是要提供一种新的雄甾-2-烯-17-酮的制备方法,以大幅降低雄甾-2-烯-17-酮的生产成本。
本发明的技术方案是,一种雄甾-2-烯-17-酮的制备方法,所述方法包括先以4-雄烯二酮为原料,经保护反应、还原反应、脱保护反应三步反应合成表雄酮,再在有机溶剂中,通过浸有酸的载体固相酸催化剂催化,将表雄酮直接脱水而脱去其分子中的3位羟基,得到目标产物雄甾-2-烯-17-酮。
进一步地,所述方法具体合成操作步骤如下:
a、固相酸催化剂的制备:是将强酸溶解于水中,加入有强吸附能力的惰性固相载体粉末,于10~50℃搅拌吸附反应3~4小时,反应完后,减压蒸去水至近干,取出干燥得固相酸催化剂,水分含量3~8%;
b、4-雄烯二酮制备表雄酮:先以4-雄烯二酮为原料,经醚化保护反应、钯炭催化加氢还原反应、酸催化水解脱保护反应三步反应合成表雄酮;步骤a和b的顺序可以互换;
c、雄甾-2-烯-17-酮的制备:是将步骤b制备的表雄酮溶解于有机溶剂中,加入上述步骤a所制备的固相酸催化剂,在30~120℃搅拌反应10~18小时,tlc确认反应终点,反应完后,趁热氮气压滤,滤饼用适量反应溶剂洗涤,然后回收固相酸催化剂套用;滤液与洗液合并,减压浓缩回收90~95%有机溶剂,然后降温,加入纯水水析,离心,滤液排入废水处理池,滤饼水洗干燥,得雄甾-2-烯-17-酮的粗品,hplc含量90.0~95.0%;粗品经丙酮与二氯甲烷混合溶剂重结晶二次,得雄甾-2-烯-17-酮精品,hplc含量97~98.5%。
进一步地,所述雄甾-2-烯-17-酮的制备方法还包括采用营养培养基和一种或多种微生物菌种使得植物甾醇经微生物发酵后制备得到4-雄烯二酮的过程。
进一步地,所述雄甾-2-烯-17-酮的制备方法还包括采用大豆油生产过程中的脱臭馏出物经包含结晶分离的步骤得到植物甾醇的过程。
进一步地,所述固相载体是氧化铝、硅胶、硅藻土、粉末硫酸镁中的一种;所用的酸催化剂是硝酸、硫酸、磷酸、醋酸、对甲苯磺酸、草酸中的一种;反应物间的重量配比是,固相载体:水:酸=1g:4~8ml:0.02~0.08g。
进一步地,所述固相载体是硅胶;所用的酸催化剂是硫酸或对甲苯磺酸;步骤a的反应温度是30~35℃;反应物间的重量配比是,固相载体:水:酸=1g:6ml:0.05g。
进一步地,所述步骤c的有机溶剂是甲苯、氯仿、乙酸乙酯、四氢呋喃、dme、dmf中的一种;反应物间的重量配比是,表雄酮:有机溶剂=1g:8~20ml,雄甾-2-烯-17-酮粗品重结晶中粗品与溶剂的配比是,雄甾-2-烯-17-酮粗品:二氯甲烷:丙酮=1g:4~8ml:2~6ml。
进一步地,所述步骤c的有机溶剂是甲苯或dmf,步骤c的反应温度是70~80℃,反应物间的重量配比是,表雄酮:固相酸催化剂=1g:3g;反应物与溶剂间的配比是,表雄酮:有机溶剂=1g:15ml;雄甾-2-烯-17-酮粗品重结晶中粗品与溶剂的配比是,雄甾-2-烯-17-酮粗品:二氯甲烷:丙酮=1g:6ml:4ml。
进一步地,先以4-雄烯二酮为原料,与原甲酸三乙酯反应生成3位得到醚化保护的醚化物,所述醚化物经钯炭催化加氢还原反应得到氢化物,所述氢化物经酸催化水解脱保护反应得到所述表雄酮。
进一步地,氢化物经酸催化水解脱保护反应过程中所用的酸催化剂为氢溴酸。
本发明的有益效果包括:
本发明以4-雄烯二酮为原料,经保护反应、还原反应、脱保护反应三步反应合成表雄酮,在有机溶剂中,通过浸有酸的载体固相酸催化剂催化,将表雄酮一步直接脱水消除其分子中的3位羟基,得到目标产物雄甾-2-烯-17-酮。
本发明合成雄甾-2-烯-17-酮的方法,相对传统合成雄甾-2-烯-17-酮方法,以从大豆油脱臭馏出物中提取的植物甾醇,经现代生物发酵技术,得到的4-雄烯二酮为原料;4-雄烯二酮经保护反应、还原反应、脱保护反应三步反应合成关键中间体表雄酮,该原料来源丰富、价格便宜。
关键中间体表雄酮制备目标产物雄甾-2-烯-17-酮,相对传统合成雄甾-2-烯-17-酮方法,合成路线由两步缩减为一步反应,经济环保,生产操作也更简便,更重要的是,采用浸有酸的载体固相酸催化剂催化,反应底物与酸催化剂非直接接触,反应温和平稳进行,脱水消除反应按热力学稳定的2,3位方向进行,反应选择性好,避免了传统碱催化消除反应时,副反应大,杂质多,杂质难精制,合成收率低,生产成本高等诸多缺陷。
以本发明方法合成雄甾-2-烯-17-酮,产品质量好,比传统的生产方法产品总收率提高25~30%,生产成本降低30~40%。工艺中使用的溶剂,可回收循环套用,既经济又环保,十分利于工业化生产。
附图说明
图1为传统表雄酮的合成路线图。
图2为传统表雄酮生产雄甾-2-烯-17-酮的合成路线图。
图3为本发明中4-雄烯二酮生产雄甾-2-烯-17-酮的合成路线图。
具体实施方式
为了更详细地说明本发明的要点与精神,下面举几个实施例予以说明。
实施例1
a、固相酸催化剂的制备:
在一个1000ml三口瓶中,加入100g固体硅胶,400ml纯水,200ml纯水与5g对甲苯磺酸配成的溶液,于30~35℃搅拌吸附反应3~4小时,反应完后,减压蒸去水至近干,取出干燥得固相酸催化剂99.8g,水分含量2.6%,重量收率99.8%;
b、4-雄烯二酮制备表雄酮:
如图3所示,以4-雄烯二酮为原料,经醚化保护反应、钯炭催化加氢还原反应、酸催化水解脱保护反应三步反应合成表雄酮。具体的,先以4-雄烯二酮为原料,与原甲酸三乙酯反应生成3位得到醚化保护的醚化物,所述醚化物经钯炭催化加氢还原反应得到氢化物,所述氢化物经酸催化水解脱保护反应得到所述表雄酮,优选最后一步中的酸催化剂为氢溴酸。
c、雄甾-2-烯-17-酮的制备:
在一个2000ml三口瓶中,加入100g表雄酮,1500ml甲苯,搅拌下使其完全溶解,再加入30g上述a所制备的固相酸催化剂,保温于70~75℃搅拌反应12~14小时,tlc确认反应终点,反应完后,趁热氮气压滤,滤饼用500ml甲苯分两次泡洗,然后回收固相酸催化剂套用;滤液与洗液合并,减压浓缩回收95%甲苯,然后降温,加入纯水水析,过滤,滤液先减压回收残余的甲苯,再排入废水池处理,滤饼水洗干燥,得雄甾-2-烯-17-酮的粗品96.5g,hplc含量93.6%;将上述方法所制备的雄甾-2-烯-17-酮粗品100g加入到600ml二氯甲烷和400ml丙酮的混合溶剂中,加热使固体完全溶解,然后常压浓缩回收约四分之三的二氯甲烷和丙酮的混合溶剂(可套用于本步精制工艺),残液慢慢冷却至-5~0℃,搅拌结晶4~5小时,过滤,滤液回收酒精与母液料套用于下批精制工艺中;滤饼用上述重结晶方法再重结晶一次,所得滤饼洗涤,烘干,得雄甾-2-烯-17-酮精品77.6g,hplc含量99.4%,精制重量收率77.6%,制备重量总收率74.9%。
实施例2
a、固相酸催化剂的制备:
在一个1000ml三口瓶中,加入100g固体氧化铝,400ml纯水,200ml纯水与5g硫酸配成的溶液,于30~35℃搅拌吸附反应3~4小时,反应完后,减压蒸去水至近干,取出干燥得固相碱催化剂99.6g,水分含量2.8%,重量收率99.6%;
b、4-雄烯二酮制备表雄酮:
如图3所示,以4-雄烯二酮为原料,经醚化保护反应、钯炭催化加氢还原反应、酸催化水解脱保护反应三步反应合成表雄酮。具体的,先以4-雄烯二酮为原料,与原甲酸三乙酯反应生成3位得到醚化保护的醚化物,所述醚化物经钯炭催化加氢还原反应得到氢化物,所述氢化物经酸催化水解脱保护反应得到所述表雄酮,优选最后一步中的酸催化剂为氢溴酸。
c、雄甾-2-烯-17-酮的制备:
在一个2000ml三口瓶中,加入100g表雄酮,1500ml氯仿,搅拌下使其完全溶解,再加入30g上述a所制备的固相酸催化剂,保温于60~65℃搅拌回流反应16~18小时,tlc确认反应终点,反应完后,趁热氮气压滤,滤饼用500ml氯仿分两次泡洗,然后回收固相酸催化剂套用;滤液与洗液合并,减压浓缩回收95%氯仿,然后降温,加入纯水水析,过滤,滤液先减压回收残余的氯仿,再排入废水处理池,滤饼水洗干燥,得雄甾-2-烯-17-酮的粗品97.2g,hplc含量91.2%;将上述方法所制备的雄甾-2-烯-17-酮粗品100g按上述实施例1所述粗品重结晶的方法将所制得雄甾-2-烯-17-酮粗品重结晶两次,得雄甾-2-烯-17-酮精品75.6g,hplc含量99.2%,精制重量收率75.6%,制备重量总收率73.5%。
实施例3
a、固相酸催化剂的制备:
在一个1000ml三口瓶中,加入100g固体硫酸镁粉末,400ml纯水,200ml纯水与5g硝酸配成的溶液,于30~35℃搅拌吸附反应3~4小时,反应完后,减压蒸去水至近干,取出干燥得固相酸催化剂99.8g,水分含量2.1%,重量收率99.8%;
b、4-雄烯二酮制备表雄酮:
如图3所示,以4-雄烯二酮为原料,经醚化保护反应、钯炭催化加氢还原反应、酸催化水解脱保护反应三步反应合成表雄酮。具体的,先以4-雄烯二酮为原料,与原甲酸三乙酯反应生成3位得到醚化保护的醚化物,所述醚化物经钯炭催化加氢还原反应得到氢化物,所述氢化物经酸催化水解脱保护反应得到所述表雄酮,优选最后一步中的酸催化剂为氢溴酸。
c、雄甾-2-烯-17-酮的制备:
在一个2000ml三口瓶中,加入100g表雄酮,1500mldmf,搅拌下使其完全溶解,再加入30g上述a所制备的固相酸催化剂,保温于75~80℃搅拌反应10~12小时,tlc确认反应终点,反应完后,趁热氮气压滤,滤饼用500mldmf分两次泡洗,然后回收固相酸催化剂套用;滤液与洗液合并,减压浓缩回收95%dmf,然后降温,加入纯水水析,过滤,滤液先减压回收残余的dmf,再排入废水处理池,滤饼水洗干燥,得雄甾-2-烯-17-酮的粗品97.4g,hplc含量90.6%;将上述方法所制备的雄甾-2-烯-17-酮粗品100g按上述实施例1所述粗品重结晶的方法将所制得雄甾-2-烯-17-酮粗品重结晶两次,得雄甾-2-烯-17-酮精品74.8g,hplc含量99.1%,精制重量收率74.8%,制备重量总收率72.9%。
以上内容是结合具体的优选实施方式对本发明作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演和替换,都应当视为属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!