一种(1R,2S)-2-(3,4-二氟苯基)环丙胺的制备方法与流程
本发明涉及化工技术领域,尤其涉及一种(1r,2s)-2-(3,4-二氟苯基)环丙胺的制备方法。
背景技术:
(1r,2s)-2-(3,4-二氟苯基)环丙胺是阿斯利康(astrazeneca)公司开发的新型抗凝血药替格瑞洛合成的关键中间体。wo2001092263以(e)-3-(3,4-二氟苯基)-2-丙烯酸为原料,通过手性诱导合成(1r,2r)-2-(3,4-二氟苯基)-环丙烷羧酸,与亚硫酰氯反应制备酰氯,由酰氯到酰基叠氮化物,并经热分解,最后得到(1r,2s)-2-(3,4-二氟苯基)环丙胺。此反应具有很大的安全风险,极易引起爆炸。
wo2011017108采用了同样的路线,但是用(pho)2p(=o)n3替代叠氮化钠。springthorpe,brianetal等(bioorganic&medicinalchemistryletters,17(21),6013-6018;2007)同样利用酰基叠氮化物热分解(1r,2s)-2-(3,4-二氟苯基)环丙胺,只是手性诱导的试剂不一样。
wo2008018822,wo2008018823利用霍夫曼(hofmann)降解反应制备(1r,2s)-2-(3,4-二氟苯基)环丙胺,酰胺与次氯酸钠或次溴酸钠的碱溶液作用时,脱去羰基生成伯胺。
wo2011132083采用还原反式-(1r,2s)-2-(3,4-二氟苯基)-1-硝基环丙烷的方法合成(1r,2s)-2-(3,4-二氟苯基)环丙胺。
然而其手性中心是通过还原剂硼烷n,n-二乙基苯基苯胺及手性辅基(s)-(-)-α,α-二苯基-2-吡咯烷甲醇下进行手性还原得到r构型的产物。硼烷等化合物易燃易爆。
wo2012001531以3,4-二氟苯乙烯为起始原料,二氯(对-癸烯)钌(ii)二聚体和(s,s)-2,6-双(4-异丙基-2-恶唑啉-2-基)吡啶催化得到手性中心,最终通过反式-(1r,2r)-n(乙酰氧基)-2-(3,4-二氟苯基)环丙烷甲酰胺制备(1r,2s)-2-(3,4-二氟苯基)环丙胺。反应使用了非常昂贵的催化剂。
zhang,hao等以手性的环氧化合物为原料(bioorganic&medicinalchemistryletters,22(11),3598-3602;2012),制备(1r,2s)-2-(3,4-二氟苯基)环丙胺。反应步骤缩短,但是制备环氧化合物的反应用到硼酸三甲酯、硼烷二甲硫醚络合物还原体系,有恶臭,环境污染严重。
wo2013144295通过(1r,2r)-2-(3,4-二苯基)环丙烷碳酰肼与亚硝酸钠反应得到(1r,2s)-2-(3,4-二氟苯基)环丙胺,然而碳酰肼也是一种不稳定的化合物,易燃易爆。
cn102924457对工艺进行了改进,通过霍夫曼(hofmann)降解反应制备(1r,2s)-2-(3,4-二氟苯基)环丙胺。
wo2013163892n-羟基-反式–(1r,2s)-2-(3,4-二氟苯基)环丙酰胺为原料,重排制备(1r,2s)-2-(3,4-二氟苯基)环丙胺。
cn103508899以(1r,2s)-2-(3,4-二氟苯基)-1-氰基环丙烷为原料,制备(1r,2s)-2-(3,4-二氟苯基)环丙胺。
然而,这些原料很难通过简单的方法得到。cn103965059通过拆分的方法得到(1r,2s)-2-(3,4-二氟苯基)环丙胺。拆分是获得光学异构体的一种简单方法,然而拆分的收率最高只能达到50%。
hugentobler,katharinag.等(organic&biomolecularchemistry,14(34),8064-8067;2016)通过酶法构建手性中心。
采用酮还原酶,酰胺酶或脂肪酶生物催化剂,合成胺的关键中间的ee值分别在99.9%,92.5%和46.3%。然而这些水解酶反应的收率最高只能达到50%。
技术实现要素:
本发明的目的是克服现有制备(1r,2s)-2-(3,4-二氟苯基)环丙胺技术中,反应条件苛刻、反应收率低,环境污染大的难题,提供一种(1r,2s)-2-(3,4-二氟苯基)环丙胺的制备方法。
本发明的技术方案如下:
将2-(3,4-二氟苯基)环丙胺溶解于溶剂中,加入2-(3,4-二氟苯基)环丙胺质量0.1-1%的南极假丝酵母脂肪酶b(candidaantarcticalipaseb,calb),加入2-(3,4-二氟苯基)环丙胺质量0.1-1%的pd/c,加入2-(3,4-二氟苯基)环丙胺1-3倍摩尔当量的短链脂肪酸酯,升温到30-60℃,保持8-24小时,降温到25℃,过滤回收pd/c、calb,滤液旋转蒸发回收溶剂,在残余油状物中滴加质量百分浓度为1-3%的盐酸水溶液,使加入的hcl与2-(3,4-二氟苯基)环丙胺的摩尔比为1-3,在30-70℃搅拌2-8小时,在溶液中滴加1-5%质量百分浓度的naoh,控制体系ph值为8-9,加入旋转蒸发过程回收的溶剂进行萃取,分层,有机相旋转蒸发回收溶剂,得到(1r,2s)-2-(3,4-二氟苯基)环丙胺。
作为本发明的优选,所述的溶剂为环己烷、正己烷或甲苯。
作为本发明的优选,所述的短链脂肪酸酯为乙酸乙酯,乙酸叔丁酯,三氟乙酸乙酯、三氟乙酸叔丁酯、丙烯酸乙酯或丙烯酸叔丁酯,优选为三氟乙酸叔丁酯。
作为本发明的优选,所述的pd/c中pd的质量百分比含量为1-5%。
以反应式表示如下:
与现有技术相比,本发明所具有的有益效果是:
1)本发明反应过程中所有步骤的温度范围均可控制在25-70℃内,进一步的,所有步骤可在接近室温的温度范围下进行,因此本发明的制备过程条件温和,反应过程不产生对环境存在污染的副产物,为(1r,2s)-2-(3,4-二氟苯基)环丙胺的制备提供了一个新的思路。
2)本发明制备过程中的原料均安全易得,解决了现有技术中需要使用硼烷、碳酰肼等易燃易爆化合物的风险,同时避免了使用昂贵金属催化剂;本发明反应过程中的溶剂、pd/c、calb均可回收套用;实际运行成本低。
3)本发明方法转化率高、选择性达90%以上,通过使用calb和pd/c,选择适宜的溶剂和反应温度调节,克服了现有技术中使用手性拆分收率最高只能达到50%的问题,相比于现有技术,获得了显著的进步。
附图说明
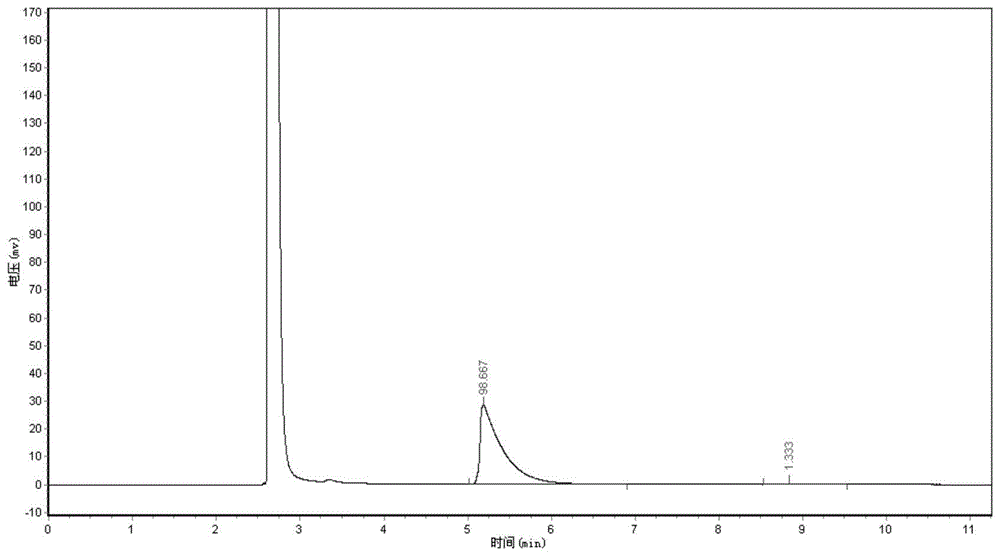
图1为实施例1中(1r,2s)-2-(3,4-二氟苯基)环丙胺的ee值测定气相色谱图。
具体实施方式
实施例1
在250毫升的锥形瓶中方法加入150毫升环己烷,0.75克2-(3,4-二氟苯基)环丙胺,0.75毫克calb,0.75毫克5%的pd/c,加入0.39克乙酸乙酯,放入摇床中,设置摇床温度为30℃,250转/分钟,反应8小时,将锥形瓶取出,降温到25℃,过滤回收pd/c、calb,滤液旋转蒸发回收溶剂145毫升,在残余油状物中滴加1%的盐酸水溶液16.2克,在30℃搅拌8小时,在溶液中滴加1%质量百分浓度的naoh,控制体系ph值为8-9,加入上述回收的环己烷145毫升萃取,分层,有机相旋转蒸发回收环己烷,得到(1r,2s)-2-(3,4-二氟苯基)环丙胺0.56克,手性气相色谱柱检测分析。气相色谱仪:shimadzugc-2014c;气相条件:色谱柱:0.25mm×25mlipodex-e;进样器温度:220℃;检测器温度:230℃;柱温:初始温度100℃,保留2min,以20℃min-1的速率升温至220℃,保留3min。测得ee值为98.6%(附图1)。比旋光度[α]=-105.0°,测试条件:质量浓度10mg/ml,溶剂为chcl3,温度20℃,钠灯589nm,旋光管长度为10cm)。
1hnmr(500mhz,cdcl3),δ:7.03-7.00(m,1h),6.80-6.73(m,2h),2.50-2.46(m,1h),1.85-1.79(m,1h),1.71(s,2h),1.08-1.05(m,1h),0.92-0.90(m,1h)。
esi-ms:170.0(m+1)+
实施例2
在250毫升的锥形瓶中方法加入150毫升正己烷,0.75克2-(3,4-二氟苯基)环丙胺,7.5毫克calb,7.5毫克1%的pd/c,加入1.54克乙酸叔丁酯,放入摇床中,设置摇床温度为60℃,250转/分钟,反应24小时,将锥形瓶取出,降温到25℃,过滤回收pd/c、calb,滤液旋转蒸发回收正己烷140毫升,在残余油状物中滴加3%的盐酸水溶液16.2克,在70℃搅拌2小时,在溶液中滴加1%质量百分浓度的naoh,控制体系ph值为8-9,加入上述回收的正己烷140毫升萃取,分层,有机相旋转蒸发回收正己烷,得到(1r,2s)-2-(3,4-二氟苯基)环丙胺0.63克,测得ee值为98.8%。
实施例3
在250毫升的锥形瓶中方法加入150毫升甲苯,0.75克2-(3,4-二氟苯基)环丙胺,7.5毫克calb,7.5毫克3%的pd/c,加入1.89克三氟乙酸乙酸,放入摇床中,设置摇床温度为60℃,250转/分钟,反应24小时,将锥形瓶取出,降温到25℃,过滤回收pd/c、calb,滤液旋转蒸发回收甲苯145毫升,在残余油状物中滴加3%的盐酸水溶液16.2克,在50℃搅拌2小时,在溶液中滴加1%质量百分浓度的naoh,控制体系ph值为8-9,加入上述回收的甲苯140毫升萃取,分层,有机相旋转蒸发回收甲苯,得到(1r,2s)-2-(3,4-二氟苯基)环丙胺0.63克,测得ee值为98.9%。
实施例4
在250毫升的锥形瓶中方法加入150毫升正己烷,0.75克2-(3,4-二氟苯基)环丙胺,7.5毫克calb,7.5毫克1%的pd/c,加入2.26克三氟乙酸叔丁酯,放入摇床中,设置摇床温度为60℃,250转/分钟,反应24小时,将锥形瓶取出,降温到25℃,过滤回收pd/c、calb,滤液旋转蒸发回收正己烷140毫升,在残余油状物中滴加3%的盐酸水溶液16.2克,在70℃搅拌2小时,在溶液中滴加1%质量百分浓度的naoh,控制体系ph值为8-9,加入上述回收的正己烷140毫升萃取,分层,有机相旋转蒸发回收正己烷,得到(1r,2s)-2-(3,4-二氟苯基)环丙胺0.72克测得ee值为99.5%。
实施例5
在250毫升的锥形瓶中方法加入150毫升环己烷,0.75克2-(3,4-二氟苯基)环丙胺,7.5毫克calb,7.5毫克1%的pd/c,加入1.33克丙烯酸乙酯,放入摇床中,设置摇床温度为60℃,250转/分钟,反应24小时,将锥形瓶取出,降温到25℃,过滤回收pd/c、calb,滤液旋转蒸发回收环己烷140毫升,在残余油状物中滴加3%的盐酸水溶液16.2克,在70℃搅拌2小时,在溶液中滴加1%质量百分浓度的naoh,控制体系ph值为8-9,加入上述回收的环己烷140毫升萃取,分层,有机相旋转蒸发回收环己烷,得到(1r,2s)-2-(3,4-二氟苯基)环丙胺0.53克,测得ee值为90.3%。
实施例6
在250毫升的锥形瓶中方法加入150毫升甲苯,0.75克2-(3,4-二氟苯基)环丙胺,7.5毫克calb,7.5毫克1%的pd/c,加入1.70克丙烯酸叔丁酯,放入摇床中,设置摇床温度为60℃,250转/分钟,反应24小时,将锥形瓶取出,降温到25℃,过滤回收pd/c、calb,滤液旋转蒸发回收甲苯140毫升,在残余油状物中滴加3%的盐酸水溶液16.2克,在70℃搅拌2小时,在溶液中滴加1%质量百分浓度的naoh,控制体系ph值为8-9,加入上述回收的甲苯140毫升萃取,分层,有机相旋转蒸发回收甲苯,得到(1r,2s)-2-(3,4-二氟苯基)环丙胺0.58克,测得ee值为93.3%。
- 还没有人留言评论。精彩留言会获得点赞!