一种亚砜参与芳基化合成多取代环1,2-二酮的制备方法与流程
本发明属于化学合成
技术领域:
,具体涉及一种亚砜参与芳基化合成多取代环1,2-二酮的制备方法。
背景技术:
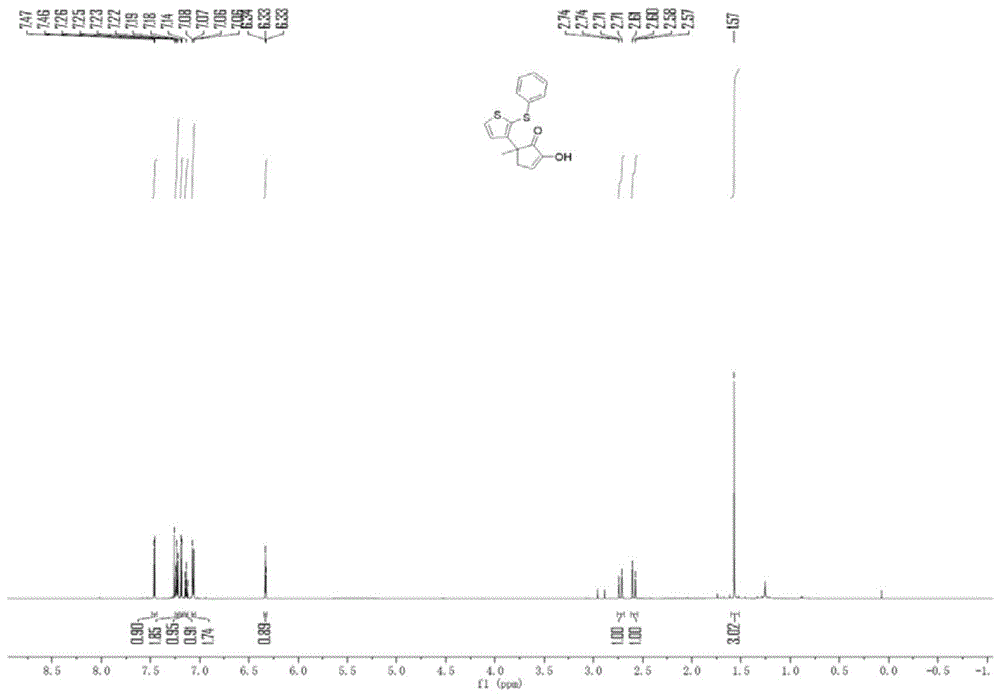
:对于合成多取代环1,2-二酮类化合物的方法,目前,主要合成方法为:由于环1,2-二酮α-C-H键的惰性和O-芳基化/烷基化的竞争,直接功能化仍然存在问题。为了获得α取代的环1,2-二酮,开发了间接方法。需要将环1,2-二酮转化为烯醇醚或烯胺,以增强此种条件下的sp2α-C-H功能化。尽管有效,这些逐步的方法通常需要使用昂贵的过渡金属和高反应温度。因此,在温和和无金属条件下,很有必要在一个步骤中实现易于接近的母体环1,2-二酮的直接区域选择性C-H功能化。技术实现要素:本部分的目的在于概述本发明的实施例的一些方面以及简要介绍一些较佳实施例。在本部分以及本申请的说明书摘要和发明名称中可能会做些简化或省略以避免使本部分、说明书摘要和发明名称的目的模糊,而这种简化或省略不能用于限制本发明的范围。鉴于上述和现有制备多取代环1,2-二酮类化合物的制备方法存在的问题,提出了本发明。因此,本发明其中的一个目的是解决现有方法中的不足,提供一种亚砜参与芳基化合成多取代环1,2-二酮类化合物的制备方法。为解决上述技术问题,本发明提供如下技术方案:一种亚砜参与芳基化合成多取代环1,2-二酮类化合物的制备方法,其包括,向试管中加入亚砜类化合物、1,2-二酮类化合物、溶剂、放在-10℃低温反应器中,待反应体系混合均匀且降为-10℃,加入三氟乙酸酐,在-10℃反应4~4.5h后移至室温,旋蒸浓缩后硅胶柱层析分离得到产物。作为本发明所述多取代环1,2-二酮类化合物的制备方法的一种优选方案:所述亚砜类化合物,其与所述1,2-二酮化合物的摩尔比为1:1~2。作为本发明所述多取代环1,2-二酮类化合物的制备方法的一种优选方案:所述三氟乙酸酐与亚砜类化合物的摩尔比为1:1~2。作为本发明所述多取代环1,2-二酮类化合物的制备方法的一种优选方案:所述溶剂的加入量为每毫摩尔亚砜类化合物对应8ml溶剂。作为本发明所述多取代环1,2-二酮类化合物的制备方法的一种优选方案:所述旋蒸,其转速为100~200rpm,温度为25~30℃,真空为0.08~0.12Mpa,处理时间为3~5min;所述层析,是采用200目柱层析硅胶,展开剂为石油醚:乙酸乙酯=9~15:1。作为本发明所述多取代环1,2-二酮类化合物的制备方法的一种优选方案:所述亚砜类化合物包括苯基、取代苯基、噻吩基亚砜化合物中的一种或几种。作为本发明所述多取代环1,2-二酮类化合物的制备方法的一种优选方案:所述1,2-二酮类化合物包括五环1,2-二酮、六环1,2-二酮中的一种或者几种。作为本发明所述多取代环1,2-二酮类化合物的制备方法的一种优选方案:所述多取代环1,2-二酮类化合物,包括式Ⅰ和Ⅱ所示多取代环1,2-二酮类化合物,式中Ar1,Ar2,Ar3,Ar4,Ar5,R2,R1为各自独立的基团,且碳数为1~12,其中n=1或2。本发明的有益效果:(1)本发明所提供的一种亚砜参与芳基化合成多取代环1,2-二酮类化合物的制备方法使得亚砜类化合物与环1,2-二酮类化合物生成C-C键。(2)整个反应无需金属催化,在有机溶剂体系,特别适用于一些药物类的合成,从根本上消除了金属残留等问题,具有较高的经济适用性。(3)操作简单,产率高,纯度在98%以上。(4)条件温和,底物范围广,不但简单底物可以适用,复杂的天然产物亦可以以此方法进行改性。附图说明为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。其中:图1为实施例1化合物的核磁谱图H谱。图2为实施例1化合物的核磁谱图C谱。图3为实施例2化合物的核磁谱图H谱。图4为实施例2化合物的核磁谱图C谱。图5为实施例3化合物的核磁谱图H谱。图6为实施例3化合物的核磁谱图C谱。图7为实施例4化合物的核磁谱图H谱。图8为实施例4化合物的核磁谱图C谱。具体实施方式为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实施例对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例的限制。其次,此处所称的“一个实施例”或“实施例”是指可包含于本发明至少一个实现方式中的特定特征、结构或特性。在本说明书中不同地方出现的“在一个实施例中”并非均指同一个实施例,也不是单独的或选择性的与其他实施例互相排斥的实施例。实施例1:向20mL的试管中,室温条件下依次加入苯基噻吩亚砜(21mg,0.1mmol)、3-甲基环戊烷-1,2二酮(17.1mg,0.15mmol),同时加入0.8ml二氯乙烷,放在-10℃低温反应器中,等反应体系混合均匀且反应体将为-10℃,加入三氟乙酸酐(21mg,0.15mmol)后,在低温-10℃下反应4h,然后使反应混合物升温至室温(通常为25℃),并在此温度下搅拌1分钟。完成后,不进行后处理操作,将试管内的反应体系转移到10ml茄形瓶内,使用Heidolph旋转蒸发仪,转速为80-100rpm,温度为38℃,真空度为0.1Mpa,处理3min,将残留物再使用200目柱层析硅胶进行柱层析,其展开剂为石油醚:乙酸乙酯=9:1,分离得目标化合物(29mg,产率为96%,经HPLC分析纯度为98%,从核磁图谱外形、信号、噪声等方面也可以反映出产品纯度极高)。1HNMR(600MHz,CDCl3)δ7.46(d,J=5.6Hz,1H),7.23(dd,J=10.9,4.6Hz,1H),7.19(d,J=5.6Hz,1H),7.14(dd,J=10.6,4.2Hz,1H),7.08–7.05(m,1H),6.33(t,J=3.1Hz,1H),2.73(dd,J=17.9,2.9Hz,1H),2.59(dd,J=17.9,3.4Hz,1H),1.57(s,1H).13CNMR(150MHz,CDCl3)δ205.80,150.35,149.75,138.33,130.18,128.88,128.39,126.61,125.96,125.74,125.51,47.32,41.23,26.11.实施例2:向20mL的试管中,室温条件下依次加入二苯基亚砜(20.6mg,0.1mmol)、3-甲基环戊烷-1,2二酮(17.1mg,0.15mmol),同时加入0.8ml二氯乙烷,放在-10℃低温反应器中,等反应体系混合均匀且反应体将为-10℃,加入三氟乙酸酐(21mg,0.15mmol)后,在低温-10℃下反应4h,然后使反应混合物升温至室温(通常为25℃),并在此温度下搅拌1分钟。完成后,不进行后处理操作,将试管内的反应体系转移到10ml茄形瓶内,使用Heidolph旋转蒸发仪,转速为80-100rpm,温度为38℃,真空度为0.1Mpa,处理3min,将残留物再使用200目柱层析硅胶进行柱层析,其展开剂为石油醚:乙酸乙酯=9:1,分离得目标化合物(26.9mg,产率为91%,经HPLC分析纯度为98%,从核磁图谱外形、信号、噪声等方面也可以反映出产品纯度极高)。1HNMR(600MHz,CDCl3)δ7.54(dd,J=8.0,1.2Hz,1H),7.39(dd,J=7.7,1.4Hz,1H),7.35–7.31(m,1H),7.26–7.20(m,3H),7.18–7.13(m,1H),7.09(dt,J=3.2,1.8Hz,2H),6.31(t,J=3.1Hz,1H),5.61(s,1H),2.76(dd,J=17.7,2.6Hz,1H),2.60(dd,J=17.7,1H),1.61(s,3H).13CNMR(150MHz,CDCl3)δ206.08,150.69,145.40,137.40,136.57,133.27,128.98,128.76,128.44,128.19,126.28,123.36,123.22,50.43,41.29,25.34.实施例3:向20mL的试管中,室温条件下依次加入4-溴苯基噻吩亚砜(30mg,0.1mmol)、1.2-环己二酮(17.1mg,0.15mmol),同时加入0.8ml二氯乙烷,放在-10℃低温反应器中,等反应体系混合均匀且反应体将为-10℃,加入三氟乙酸酐(21mg,0.15mmol)后,在低温-10℃下反应4h,然后使反应混合物升温至室温(通常为25℃),并在此温度下搅拌1分钟。完成后,不进行后处理操作,将试管内的反应体系转移到10ml茄形瓶内,使用Heidolph旋转蒸发仪,转速为80-100rpm,温度为38℃,真空度为0.1Mpa,处理3min,将残留物再使用200目柱层析硅胶进行柱层析,其展开剂为石油醚:乙酸乙酯=9:1,分离得目标化合物(22.9mg,产率为60%,经HPLC分析纯度为98%,从核磁图谱外形、信号、噪声等方面也可以反映出产品纯度极高)。1HNMR(400MHz,CDCl3)δ7.49(d,J=5.5Hz,1H),7.37–7.32(m,1H),7.22(d,J=5.5Hz,1H),7.03–6.97(m,1H),6.39(s,1H),2.75(t,J=5.9Hz,1H),2.60–2.52(m,1H),2.07–1.97(m,1H).13CNMR(100MHz,CDCl3)δ195.12,144.06,143.77,137.66,132.02,130.03,129.62,128.52,127.69,125.20,119.86,36.05,29.45,22.94.实施例4:首先和实例1中一样的步骤,向20mL的试管中,室温条件下依次加入苯基噻吩亚砜(42mg,0.2mmol)、3-甲基环戊烷-1,2二酮(34.2mg,0.3mmol),同时加入1.6ml二氯乙烷,放在-10℃低温反应器中,等反应体系混合均匀且反应体将为-10℃,加入三氟乙酸酐(42mg,0.3mmol)后,在低温-10℃下反应4h,然后使反应混合物升温至室温(通常为25℃),并在此温度下搅拌1分钟。完成后,不进行后处理操作,将试管内的反应体系转移到25ml茄形瓶内,使用Heidolph旋转蒸发仪,转速为80-100rpm,温度为38℃,真空度为0.1Mpa,处理3min,将残留物再使用200目柱层析硅胶进行柱层析,其展开剂为石油醚:乙酸乙酯=9:1,分离得目标化合物(58mg,产率为96%,经HPLC分析纯度为98%,从核磁图谱外形、信号、噪声等方面也可以反映出产品纯度极高)。取上一步的产物(46.5mg,0.15mmol),苯基噻吩亚砜(21mg,0.1mmol),向20mL的试管中室温条件下依次加入,同时加入0.8ml二氯乙烷,放在-10℃低温反应器中,等反应体系混合均匀且反应体将为-10℃,加入三氟乙酸酐(21mg,0.15mmol)后,在低温-10℃下反应4h,然后使反应混合物升温至室温(通常为25℃),并在此温度下搅拌1分钟。完成后,不进行后处理操作,将试管内的反应体系转移到10ml茄形瓶内,使用Heidolph旋转蒸发仪,转速为80-100rpm,温度为38℃,真空度为0.1Mpa,处理3min,将残留物再使用200目柱层析硅胶进行柱层析,其展开剂为石油醚:乙酸乙酯=9:1,分离得目标化合物(32.1mg,产率为65%,经HPLC分析纯度为98%,从核磁图谱外形、信号、噪声等方面也可以反映出产品纯度极高)。1HNMR(400MHz,CDCl3)δ7.86(d,J=5.7Hz,1H),7.45(dd,J=5.6,3.7Hz,2H),7.25–6.95(m,11H),6.52(s,1H),3.42(d,J=17.3Hz,1H),3.25(d,J=17.3Hz,1H),1.52(s,3H).13CNMR(100MHz,CDCl3)δ205.24,149.94,145.87,138.91,138.53,138.11,131.56,130.55,130.30,130.11,129.46,129.19,128.77,128.62,127.06,126.66,126.29,125.91,125.44,46.52,43.61,26.23.实施例5:向20mL的试管中,室温条件下依次加入苯基噻吩亚砜(21mg,0.1mmol)、3-甲基环戊烷-1,2二酮(17.1mg,0.15mmol),同时加入表1中不同浓度的二氯乙烷,放在80℃反应器中,等反应体系混合均匀且反应体将为80℃,加入三氟乙酸酐(21mg,0.15mmol)后,在低温80℃下反应4h。溶剂浓度对产率的影响见表1表1不同溶剂浓度对产率的影响序号浓度(M=mol/L)产率(%)10.12820.1256130.155040.23650.5176112实施例6:向20mL的试管中,室温条件下依次加入苯基噻吩亚砜(21mg,0.1mmol)、3-甲基环戊烷-1,2二酮(17.1mg,0.15mmol),同时加入0.8ml二氯乙烷,放在低温反应器中,等反应体系混合均匀,加入三氟乙酸酐(21mg,0.15mmol)后,在表2中的不同温度下反应4h。表2不同反应温度对产率的影响序号温度(T/℃)产率(%)1-30772-109633067本发明所提供的一种亚砜参与芳基化合成多取代环1,2-二酮类化合物的制备方法使得亚砜类化合物与环1,2-二酮类化合物生成C-C键。整个反应无需金属催化,在有机溶剂体系,特别适用于一些药物类的合成,从根本上消除了金属残留等问题,具有较高的经济适用性。操作简单,产率高,纯度在98%以上。条件温和,底物范围广,不但简单底物可以适用,复杂的天然产物亦可以以此方法进行改性。研发制得的这类化合物既是受欢迎的中间体,也可以经过后续衍生成为药物等多种天然产物的基础骨架。应说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围当中。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1