一种连续的、低成本的伊鲁替尼的制备方法与流程
本发明涉及伊鲁替尼的制备方法,属于医药合成技术领域。
背景技术:
伊鲁替尼(ibrutinib),又名依鲁替尼化学名称(r)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1h-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one,cas号:936563-96-1,分子量440.5,属于小分子靶向药物。由美国pharmacyclics公司和强生公司联合开发,是一种口服的名为布鲁顿酪氨酸激酶(btk)抑制剂的首创新药,用于治疗慢性淋巴细胞白血病(cll)和套细胞淋巴瘤(mcl)等疾病,并于2013年11月13日获得美国食品药品管理局(fda)批准上市,该药物由于对多种疾病独特而显著的治疗效果,近年来在国际上销量增长迅速,需求量日益增多。其结构式为:
ⅰ
对于该药物的合成,国内外已有相关报道,目前主要采用的路线是以中间体3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺(化合物ⅱ)为原料和(r,s)-1-叔丁氧羰基-3-羟基哌啶(化合物ⅴ)进行mitsunobu反应后,再经脱保护、酰化制得伊鲁替尼。如us200680056438.5所述,路线如下:
该路线具有反应条件温和,反应选择性高,副反应少等优点,但该路线在mitsunobu反应时为保证反应效果,加入的diad和三苯基膦聚合物比例大,三苯基膦聚合物昂贵,使得制造成本高,且该路线在酰化反应后,采用硅胶柱层析的方法来进行纯化,耗时费力,生产效率低。
针对以上问题,cn106146512a用三苯基膦代替了三苯基膦聚合物,酰化过程采用丙烯酸酐与化合物ⅶ缩合,经过重结晶获得伊鲁替尼。该路线采用了易得的三苯基膦,且不经柱层析,一定程度简化的工艺;但diad和三苯基膦的投料摩尔比都是化合物ⅱ的3倍量,仍造成母液处理困难,产品夹带杂质多,终产品仍需进行多次重结晶才能保证质量,步骤繁琐。
cn106008525a采用了1.5~4倍的摩尔量的diad和三苯基膦进行反应,通过和丙烯酸脂在催化剂和活化剂存在的条件下反应获得伊鲁替尼。但该路线的同样存在diad和三苯基膦用量偏大的问题,同时最终产物依鲁替尼的杂质也较多。
cn106083860a提供了一种3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺:(s)-1-叔丁氧羰基-3-羟基哌啶:diad:三苯基膦=1:1:1:1的投料方式,反应得到的伊鲁替尼中间体式ⅶ采用甲醇乙酸乙酯混合溶剂重结晶获得高纯度的精品。虽然该法diad和三苯基膦投料用量少,但原料3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺残留量大,后处理没有有效的去除方法,只能通过重结晶来提高纯度,操作繁琐;由此也造成产品收率低,间接提高了生产成本。
从以上专利方法可知,在该合成路线mitsunobu反应中,diad和tpp的用量少时,反应体系会残留大量未反应的3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺,形成原料浪费且目前缺少较好的去除手段,只能采用重结晶的方式纯化;diad和tpp投料的用量多时,反应完成会生成大量的三苯基氧磷等三废,既造成了生成成本的急剧增加,也使后处理难度增加,也需要结晶中间体式ⅶ来精制,工序繁琐。
技术实现要素:
针对现有技术中存在的技术问题,本发明要解决的技术问题在于,克服现有技术中原料残留量大、三苯基氧磷使用过量、成本高等缺陷;提供一种综合收率高、生产成本低,环境友好连续的伊鲁替尼的制备方法,所得伊鲁替尼hplc纯度≥99﹪,适合工业化生产。
连续的、低成本的伊鲁替尼的制备方法,包括以下步骤:
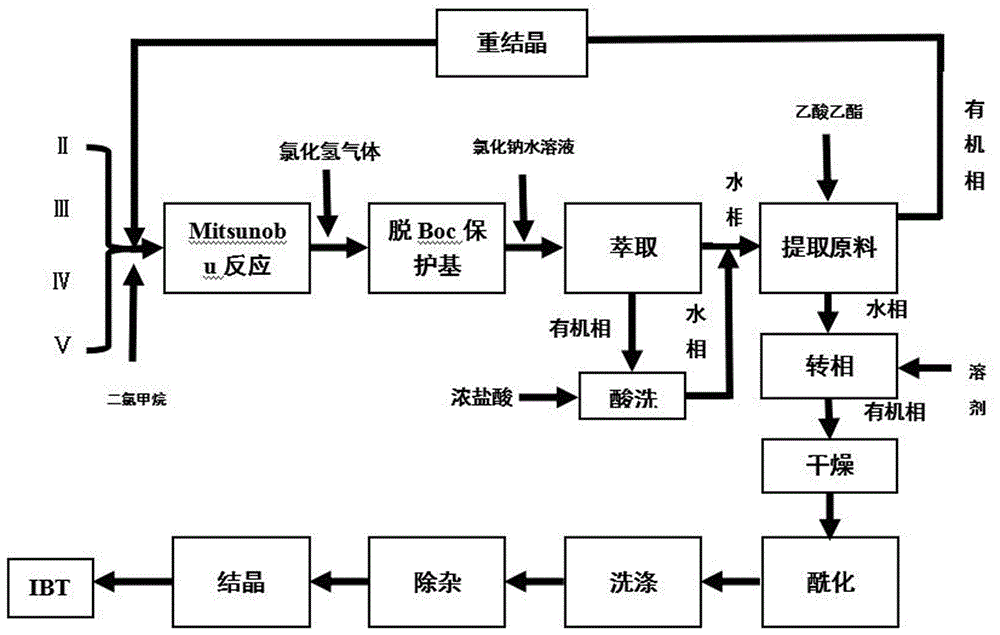
1)3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺与(s)-1-叔丁氧羰基-3-羟基哌啶、diad、三苯基膦在溶剂二氯甲烷中进行mitsunobu反应,获得中间体式ⅵ(r)-3-[4-氨基-3-(4-苯氧基苯基)-1h-吡唑并[3,4-d]嘧啶-1-基]哌啶-1-羧酸叔丁酯;
3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺与(s)-1-叔丁氧羰基-3-羟基哌啶、diad、三苯基膦的投料摩尔比为1:1-1.5:0.7-1.5:0.7-1.5;
2)反应液直接通入氯化氢气体脱boc保护基至反应液由澄清变混浊,加入氯化钠水溶液萃取,萃取后的有机相再加入浓盐酸提取未反应的3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺,浓盐酸用量(体积)应大于有机相体积的一半,合并水相;用氢氧化钠溶液调节ph,再用乙酸乙酯提取水相中3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺,蒸除有机相溶剂后,加入重结晶溶剂进行精制,分离干燥,所得3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺进行投料套用;
3)步骤2)提取原料后的水相中加入二氯甲烷,再用氢氧化钠水溶液调ph,分出有机相,用无水硫酸钠干燥后,直接加入三乙胺和丙烯酰氯进行酰化反应;
4)反应完成后的料液依次用柠檬酸水溶液、饱和食盐水洗涤,之后加入硅胶和活性炭搅拌除杂,过滤后蒸除溶剂,得hplc纯度≥99﹪的伊鲁替尼。
优选的步骤1)中3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺与(s)-1-叔丁氧羰基-3-羟基哌啶、diad、三苯基膦的投料摩尔比为1:1.3:1.3:1.3。
步骤2)中,水相用氢氧化钠水溶液调节ph至1.8-3.5;提取后的原料的重结晶溶剂选自甲醇、乙醇、异丙醇、乙腈或水中的一种或多种。
优选的步骤2)中水相调节ph为2.8;重结晶溶剂为甲醇和水的混合物,甲醇与水的体积比为2:3。
步骤3)中调节ph至10-11;优选为ph=11。
步骤4)中,反应液加入硅胶和活性炭的质量比为1:1,质量各为3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺投料质量的5﹪-10﹪。
通过实验发现了脱boc后的反应料液通过不同ph条件下、用不同溶剂洗涤萃取的方法能够实现杂质、主原料3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺和中间体的良好分离,回收后的主原料可直接套用,所得中间体不经结晶可直接用于下一步反应,并且反应杂质少,不需要用柱层析的方法进行纯化,仅用少量硅胶和活性炭搅拌除杂,就可以较高的综合收率获得较高质量的伊鲁替尼。
本发明利用乙酸乙酯对主原料和中间产物的溶解度差异来实现不同物料的分离,分离效果好。
步骤2)反应液直接通入氯化氢气体脱boc保护基:体系内滴加等物质的量的盐酸和通入等物质的量的氯化氢气体相比,氯化氢气体可以使反应更彻底,而如果要达到相同的反应效果,则需要更多量的浓盐酸。
本发明的创新之处在于:
①通过精准控制ph值且用乙酸乙酯洗涤达到完美分离主原料和伊鲁替尼中间体的效果;
②本发明采用连续法工艺,无需结晶和重结晶,既工艺简单,亦能保证产品纯度;
③本发明采用极低的物料配比,减少辅料用量,环境友好;但回收套用未反应主原料,综合套用收率较高,成本低廉,有利于工业化生产。
本发明的有益效果在于:
1.三苯基膦和diad的投料比例小,既降低了后处理过程产物的提纯难度,又减少了工业三废的产生,环境友好;
2.反应过程中避免了多次溶剂置换步骤,溶剂回收套用方便,节约能耗,生产效率高;
3.未反应原料直接回收套用,综合收率高,生产成本低;
4.整个反应连续操作,中间过程无结晶和柱层析等步骤,无需多次重结晶,操作简便,利于工业化生产。
附图说明
图1为本发明的工艺流程图。
具体实施方式
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
实施例1
向100ml的反应瓶中加入40ml二氯甲烷,搅拌下依次加入(2.875g,0.0095mol)3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺、(2.479g,0.0123mol)(s)-1-叔丁氧羰基-3-羟基哌啶、(2.479g,0.0123mol)三苯基膦。控温25℃下,滴加偶氮二甲酸二异丙酯的二氯甲烷溶液(将2.492g,0.0123mol的diad溶于15ml二氯甲烷中),35min滴毕,保温反应5h。
控温0℃,搅拌下往反应液中通入hcl气体,持续4h,之后保温反应3h后加入氯化钠水溶液萃取,二氯相(萃取后得到的有机相)搅拌下加入30ml浓盐酸,搅拌30min后分离水相并使两次水相合并,搅拌下用10﹪的氢氧化钠水溶液调ph=2.8,加入60ml乙酸乙酯,萃取化合物式ⅱ。水相再用60ml乙酸乙酯洗涤两次,加入40ml二氯甲烷,用10﹪氢氧化钠水溶液调ph=11,分液,保留二氯甲烷相,加入2.0g无水硫酸钠干燥后,准备用于下一步反应。
乙酸乙酯相合并后蒸除溶剂,用甲醇水重结晶(甲醇:水=2:3),干燥得3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺0.358g,hplc纯度99.38﹪。
取上一步得到的反应液,加入(1.693g,0.0167mol)三乙胺,控温-5℃,搅拌下滴加丙烯酰氯的二氯甲烷溶液(将0.606g,0.0066mol的丙烯酰氯溶于10ml的二氯甲烷中),30min滴毕。保温反应3h。用5﹪的柠檬酸水溶液各60ml洗涤三次。加入0.21g硅胶和0.21g活性炭搅拌3h除杂,过滤,60ml饱和食盐水洗涤一次,1.0g无水硫酸钠干燥,蒸除溶剂,得伊鲁替尼1.655g,以3-(4-苯氧基苯基)-1h-吡唑[3,4-d]嘧啶-4-胺实际消耗为准,收率为65.79﹪,hplc纯度99.56﹪。
实施例2:
向100ml的反应瓶中加入40ml二氯甲烷,搅拌下依次加入(2.875g,0.0095mol)化合物式ⅱ(含实施例1回收化合物式ⅱ0.358g)、(1.907g,0.0095mol)化合物式ⅴ、(1.989g,0.0076mol)化合物式ⅲ。控温25℃下,滴加化合物式ⅳ的二氯甲烷溶液(将1.533g,0.0076mol的diad溶于15ml二氯甲烷中),35min滴毕,保温反应5h。
控温0℃,搅拌下往反应液中通入hcl气体,持续4h。保温反应3h。加入氯化钠水溶液萃取。二氯相搅拌下加入30ml浓盐酸,搅拌30min后分离水相并使两次水相合并,搅拌下用10﹪的氢氧化钠水溶液调ph=3.1,加入60ml乙酸乙酯,萃取化合物式ⅱ。水相再用60ml乙酸乙酯洗涤两次,加入40ml二氯甲烷,用10﹪氢氧化钠水溶液调ph=11,分液,保留二氯甲烷相,加入2.0g无水硫酸钠干燥后,准备用于下一步反应。
乙酸乙酯相合并后蒸除溶剂,用乙醇水重结晶(乙醇:水=1:2),干燥得化合物式ⅱ1.014g,hplc纯度99.15﹪。
取上一步得到的反应液,加入(1.037g,0.010mol)三乙胺,控温-5℃,搅拌下滴加丙烯酰氯的二氯甲烷溶液(将0.371g,0.0041mol的丙烯酰氯溶于10ml的二氯甲烷中),30min滴毕。保温反应3h。用5﹪的柠檬酸水溶液各60ml洗涤三次。加入0.20g硅胶和0.20g活性炭搅拌3h除杂,过滤,60ml饱和食盐水洗涤一次,1.0g无水硫酸钠干燥,蒸除溶剂,得伊鲁替尼0.994g,以化合物式ⅱ实际消耗为准,收率为54.16﹪,hplc纯度99.34﹪。
实施例3:
向100ml的反应瓶中加入40ml二氯甲烷,搅拌下依次加入(2.875g,0.0095mol)化合物式ⅱ(含实施例2回收化合物式ⅱ1.014g)、(2.479g,0.0123mol)化合物式ⅴ、(3.232g,0.0123mol)化合物式ⅲ。控温20℃下,滴加化合物式ⅳ的二氯甲烷溶液(将2.492g,0.0123mol的diad溶于15ml二氯甲烷中),35min滴毕,保温反应5h。
控温0℃,搅拌下往反应液中通入hcl气体,持续4h。保温反应3h。加入氯化钠水溶液萃取,二氯相搅拌下加入30ml浓盐酸,搅拌30min后分离水相并使两次水相合并,搅拌下用10﹪的氢氧化钠水溶液调ph=2.5,加入60ml乙酸乙酯,萃取化合物式ⅱ。水相再用60ml乙酸乙酯洗涤两次,加入40ml二氯甲烷,用10﹪氢氧化钠水溶液调ph=11,分液,保留二氯甲烷相,加入2.0g无水硫酸钠干燥后,准备用于下一步反应。
乙酸乙酯相合并后蒸除溶剂,用甲醇水重结晶(甲醇:水=2:3),干燥得化合物式ⅱ0.478g,hplc纯度99.11﹪。
取上一步得到的反应液,加入(1.544g,0.0152mol)三乙胺,控温-5℃,搅拌下滴加丙烯酰氯的二氯甲烷溶液(将0.552g,0.0061mol的丙烯酰氯溶于10ml的二氯甲烷中),30min滴毕。保温反应3h。用5﹪的柠檬酸水溶液各60ml洗涤三次。加入0.19g硅胶和0.19g活性炭搅拌3h除杂,过滤,60ml饱和食盐水洗涤一次,1.0g无水硫酸钠干燥,蒸除溶剂,得伊鲁替尼1.538g,以化合物式ⅱ实际消耗为准,收率为64.20﹪,hplc纯度99.29﹪。
实施例4:
向100ml的反应瓶中加入40ml二氯甲烷,搅拌下依次加入(2.875g,0.0095mol)化合物式ⅱ(含实施例3回收化合物式ⅱ0.478g)、(2.479g,0.0123mol)化合物式ⅴ、(3.232g,0.0123mol)化合物式ⅲ。控温20℃下,滴加化合物式ⅳ的二氯甲烷溶液(将2.492g,0.0123mol的diad溶于15ml二氯甲烷中),35min滴毕,保温反应5h。
控温5℃,搅拌下往反应液中通入hcl气体,持续4h。保温反应3h。加入氯化钠水溶液萃取。二氯相搅拌下加入30ml浓盐酸,搅拌30min后分离水相并使两次水相合并,搅拌下用10﹪的氢氧化钠水溶液调ph=2.6,加入60ml乙酸乙酯,萃取化合物式ⅱ。水相再用60ml乙酸乙酯洗涤两次,加入40ml二氯甲烷,用10﹪氢氧化钠水溶液调ph=11,分液,保留二氯甲烷相,加入2.0g无水硫酸钠干燥后,准备用于下一步反应。
乙酸乙酯相合并后蒸除溶剂,用甲醇水重结晶(甲醇:水=2:3),干燥得化合物式ⅱ0.491g,hplc纯度99.35﹪。
取上一步得到的反应液,加入(1.543g,0.0152mol)三乙胺,控温-5℃,搅拌下滴加丙烯酰氯的二氯甲烷溶液(将0.552g,0.0061mol的丙烯酰氯溶于10ml的二氯甲烷中),30min滴毕。保温反应3h。用5﹪的柠檬酸水溶液各60ml洗涤三次。加入0.22g硅胶和0.22g活性炭搅拌3h除杂,过滤,60ml饱和食盐水洗涤一次,1.0g无水硫酸钠干燥,蒸除溶剂,得伊鲁替尼1.574g,以化合物式ⅱ实际消耗为准,收率为66.15﹪,hplc纯度99.18﹪。
实施例5:
向100ml的反应瓶中加入40ml二氯甲烷,搅拌下依次加入(2.875g,0.0095mol)化合物式ⅱ(含实施例4回收化合物式ⅱ0.491g)、(2.864g,0.0142mol)化合物式ⅴ、(3.232g,0.0123mol)化合物式ⅲ。控温20℃下,滴加化合物式ⅳ的二氯甲烷溶液(将3.729g,0.0142mol的diad溶于15ml二氯甲烷中),35min滴毕,保温反应5h。
控温0℃,搅拌下往反应液中通入hcl气体,持续4h。保温反应3h。加入氯化钠水溶液萃取。二氯相搅拌下加入30ml浓盐酸,搅拌30min后分离水相并使两次水相合并,搅拌下用10﹪的氢氧化钠水溶液调ph=3.0,加入60ml乙酸乙酯,萃取化合物式ⅱ。水相再用60ml乙酸乙酯洗涤两次,加入40ml二氯甲烷,用10﹪氢氧化钠水溶液调ph=11,分液,保留二氯甲烷相,加入2.0g无水硫酸钠干燥后,准备用于下一步反应。
乙酸乙酯相合并后蒸除溶剂,用乙醇水重结晶(乙醇:水=1:2),干燥得化合物式ⅱ0.450g,hplc纯度98.68﹪。
取上一步得到的反应液,加入(1.553g,0.0153mol)三乙胺,控温-5℃,搅拌下滴加丙烯酰氯的二氯甲烷溶液(将0.556g,0.0061mol的丙烯酰氯溶于10ml的二氯甲烷中),30min滴毕。保温反应3h。用5﹪的柠檬酸水溶液各60ml洗涤三次。加入0.21g硅胶和0.21g活性炭搅拌3h除杂,过滤,60ml饱和食盐水洗涤一次,1.0g无水硫酸钠干燥,蒸除溶剂,得伊鲁替尼1.560g,以化合物式ⅱ实际消耗为准,收率为64.35﹪,hplc纯度99.27﹪。
实施例6:
向100ml的反应瓶中加入40ml二氯甲烷,搅拌下依次加入(2.875g,0.0095mol)化合物式ⅱ(含实施例4回收化合物式ⅱ0.450g)、(2.864g,0.0142mol)化合物式ⅴ、(3.232g,0.0123mol)化合物式ⅲ。控温20℃下,滴加化合物式ⅳ的二氯甲烷溶液(将3.729g,0.0142mol的diad溶于15ml二氯甲烷中),35min滴毕,保温反应5h。
控温0℃,搅拌下往反应液中通入hcl气体,持续4h。保温反应3h。加入氯化钠水溶液萃取。二氯相搅拌下加入30ml浓盐酸,搅拌30min后分离水相并使两次水相合并,搅拌下用10﹪的氢氧化钠水溶液调ph=2.7,加入60ml乙酸乙酯,萃取化合物式ⅱ。水相再用60ml乙酸乙酯洗涤两次,加入40ml二氯甲烷,用10﹪氢氧化钠水溶液调ph=11,分液,保留二氯甲烷相,加入2.0g无水硫酸钠干燥后,准备用于下一步反应。
乙酸乙酯相合并后蒸除溶剂,用甲醇水重结晶(甲醇:水=1:2),干燥得化合物式ⅱ0.301g,hplc纯度99.26﹪。
取上一步得到的反应液,加入(1.708g,0.0168mol)三乙胺,控温-5℃,搅拌下滴加丙烯酰氯的二氯甲烷溶液(将0.611g,0.0067mol的丙烯酰氯溶于10ml的二氯甲烷中),30min滴毕。保温反应3h。用5﹪的柠檬酸水溶液各60ml洗涤三次。加入0.20g硅胶和0.20g活性炭搅拌3h除杂,过滤,60ml饱和食盐水洗涤一次,1.0g无水硫酸钠干燥,蒸除溶剂,得伊鲁替尼1.725g,以化合物式ⅱ实际消耗为准,收率为67.12﹪,hplc纯度99.37﹪。
实施例7:
向100ml的反应瓶中加入40ml二氯甲烷,搅拌下依次加入(2.875g,0.0095mol)化合物式ⅱ(含实施例4回收化合物式ⅱ0.301g)、(2.864g,0.0142mol)化合物式ⅴ、(3.232g,0.0123mol)化合物式ⅲ。控温20℃下,滴加化合物式ⅳ的二氯甲烷溶液(将3.729g,0.0142mol的diad溶于15ml二氯甲烷中),35min滴毕,保温反应5h。
控温0℃,搅拌下往反应液中通入hcl气体,持续4h。保温反应3h。加入氯化钠水溶液萃取。二氯相搅拌下加入30ml浓盐酸,搅拌30min后分离水相并使两次水相合并,搅拌下用10﹪的氢氧化钠水溶液调ph=3.0,加入60ml乙酸乙酯,萃取化合物式ⅱ。水相再用60ml乙酸乙酯洗涤两次,加入40ml二氯甲烷,用10﹪氢氧化钠水溶液调ph=11,分液,保留二氯甲烷相,加入2.0g无水硫酸钠干燥后,准备用于下一步反应。
乙酸乙酯相合并后蒸除溶剂,用甲醇水重结晶(甲醇:水=2:3),干燥得化合物式ⅱ0.388g,hplc纯度99.43﹪。
取上一步得到的反应液,加入三乙胺(1.696g,0.0167mol),控温-5℃,搅拌下滴加丙烯酰氯的二氯甲烷溶液(将0.607g,0.0067mol的丙烯酰氯溶于10ml的二氯甲烷中),30min滴毕。保温反应3h。用5﹪的柠檬酸水溶液各60ml洗涤三次。加入0.23g硅胶和0.23g活性炭搅拌3h除杂,过滤,60ml饱和食盐水洗涤一次,1.0g无水硫酸钠干燥,蒸除溶剂,得伊鲁替尼1.667g,以化合物式ⅱ实际消耗为准,收率为67.04﹪,hplc纯度99.08﹪。
申请人声明,本发明通过上述实施例来说明本发明,但本发明并不局限于上述工艺步骤,即不意味着本发明必须依赖上述工艺步骤才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!