一种异更昔洛韦的制备方法与流程
1.本发明属于医药技术领域,具体涉及抗病毒药物更昔洛韦有关物质异更昔洛韦的制备方法。
背景技术:
2.更昔洛韦(ganciclovir)化学名为9-(1,3-二羟基-2-丙氧甲基)鸟嘌呤,是美国syntex公司研发的核苷类抗病毒药,1988年6月首次于英国上市。更昔洛韦能有效抑制巨细胞病毒和疱疹病毒,是目前临床上广泛应用的抗巨细胞病毒感染药物之一,但由于口服生物利用度差,主要通过注射给药。盐酸缬更昔洛韦是更昔洛韦的缬氨酸酯前药,口服生物利用度大大改善,进入人体后,在肠道和肝脏细胞中迅速被酯酶水解分离出缬氨酸和更昔洛韦,目前在欧美已逐渐取代更昔洛韦成为抗巨细胞病毒的一线药物。
3.异更昔洛韦化学名为9-(2,3-二羟基-1-丙氧甲基)鸟嘌呤,是美国药典中抗病毒药更昔洛韦的杂质a,同时异更昔洛韦也用于合成美国药典中缬更昔洛韦杂质d(即异缬更昔洛韦)。异更昔洛韦的结构式如下:
[0004][0005]
现有文献报道的异更昔洛韦的合成方法主要有以下几种:
[0006]
方法一:文献synthesis of 9-(2,3-dihydroxy-1-propoxymethyl)guanine-a new potential antiviral agent[j].tetrahedron letters,1984,25:905-906,tai-shun lin,mao-chin liu报道了如下合成路线
[0007]
[0008]
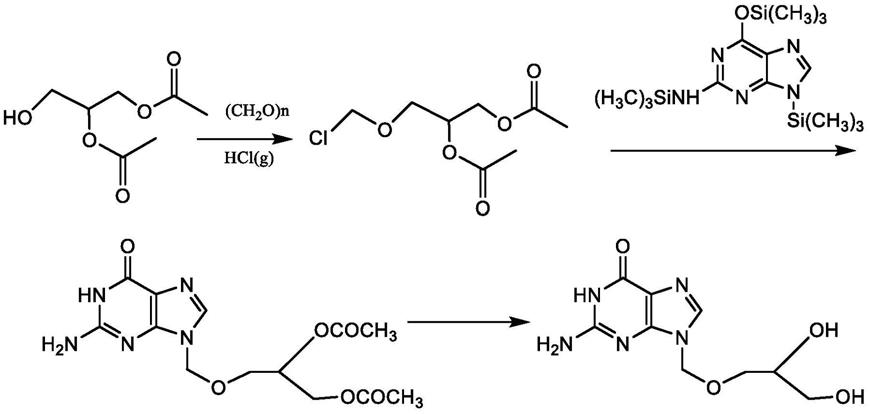
这条反应路线是通过三甲基硅保护的鸟嘌呤和氯甲基醚侧链的缩合反应和之后的脱保护基得到异更昔洛韦,合成路线较简便。但制备氯甲基醚侧链的原料1,2-二乙酰氧基-3-丙醇较难获得,其中会含有较多的1,3-二乙酰氧基-2-丙醇杂质,用其制备异更昔洛韦较难获得较高的纯度。
[0009]
方法2:文献synthesis of valganciclovir hydrochloride congeners[j].synth.commen.,2013,43:1751-1758,babu k s,rao m r,goverdhan g报道路线如下:
[0010][0011]
该路线以烯丙醇为原料,通过羟基的氯甲基醚化得到3-氯甲氧基-1-丙烯,然后与6-氧苄基鸟嘌呤进行缩合反应,而后经过烯烃双键氧化、氢解脱保护基得到异更昔洛韦。文献没有给出每步的收率,也没有具体的实验步骤。方法2合成3-氯甲氧基-1-丙烯,反应过程中氯化氢易与烯丙醇及产物氯甲基烯丙醚中的双链发生加成,产生大量副产物。南昌大学丁永翔等人重复此路线时,在第二步的缩合反应和第三步的双羟基化反应中均生成了较大杂质,难以纯化而得到纯净的相应产物。
[0012]
方法3:文献9-[(2,3-二羟基-丙氧基)甲基]鸟嘌呤的合成[j].南昌大学学报(理科版),第39卷第1期,2015年2月:66-69报道路线如下:
[0013][0014]
该路线以烯丙醇为原料,通过羟基的氯甲基醚化、乙酸钾取代、烯烃双键氧化及乙
酸酐酯化得到侧链2,3-二乙酰氧基-1-乙酰氧基甲氧基丙烷,接着与二乙酰鸟嘌呤进行缩合反应,然后经水解反应得到异更昔洛韦。
[0015]
该方法存在以下问题:据此文献报道,侧链中间体3-氯甲氧基-1-丙烯稳定性较差,在室温下易分解,较难纯化,而相关杂质的存在会影响下步乙酸钾取代反应效果,甚至可能不反应;在将3-乙酰氧基甲氧基-1-丙烯的双键氧化变成双羟基过程中,存在羟基被继续氧化的风险,副产物较多,纯化产物极为困难,文献中没有相关纯度报道。经柱层析得到的侧链2,3-二乙酰氧基-1-乙酰氧基甲氧基丙烷与二乙酰鸟嘌呤缩合得到的n
2-乙酰基-9-[(2,3-二乙酰氧基丙氧基)甲基]鸟嘌呤需要经过硅胶柱层析进行纯化处理,最终水解得到异更昔洛韦粗品,进行重结晶得到0.55g,未报道纯度。该方法操作复杂,难以批量合成异更昔洛韦。
[0016]
方法4:文献9-[(2,3-二羟基-1-丙氧基)甲基]鸟嘌呤的合成[j].南昌大学学报(理科版),第40卷第4期,2016年8月:330-340,报道路线如下:
[0017][0018]
该路线以苄基缩水甘油醚为原料,依次通过酸催化的环氧开环反应、伯羟基的选择性保护、仲羟基苄醚化反应、伯羟基去保护、氯甲基醚化和取代反应得到侧链2,3-二苄氧基-1-乙酰基甲氧基丙烷,然后2,3-二苄氧基-1-乙酰基甲氧基丙烷与二乙酰鸟嘌呤发生缩合反应,所得缩合产物再经过氢化、水解反应得到异更昔洛韦。合成的侧链中间体3-苄氧基-1,2-丙二醇、3-苄氧基-1-三苯基甲基-2-丙醇、2,3-二苄氧基-1-三苯基甲基-2-丙烷、2,3-二苄氧基-1-丙醇均需硅胶柱层析分离,操作复杂。文献报道最终产物异更昔洛韦质量所得0.4g,不能批量合成,纯度未报道。该方法的合成路线长,反应复杂,需进行氢化反应。
[0019]
方法5:文献an alternative approach to isoganciclovir:a prominent impurity in the antiviral drug ganciclovir[j].scientia pharmaceutica,2015,83:233-241,d.t.s.s.sundaram报道路线如下:
[0020][0021]
该路线以甘油、环丙酮为起始原料,生成的侧链(1,4)-二氧杂螺[4.5]癸烷-2-甲氧基)乙酸甲酯有副产物2-[(甲氧基甲氧基)甲基]-(1,4)-二氧杂螺[4.5]癸烷生成,较难纯化。侧链(1,4)-二氧杂螺[4.5]癸烷-2-甲氧基)乙酸甲酯和二乙酰鸟嘌呤发生缩合反应时,产物n
2-乙酰基-9-[(1,4)-二氧杂螺[4.5]癸烷-2-甲氧基)甲基]鸟嘌呤含有较多杂质,据文献报道有20%的n7异构体副产物n
2-乙酰基-7-[(1,4)-二氧杂螺[4.5]癸烷-2-甲氧基)甲基]鸟嘌呤产生,较难分离。方法5最终得到了异更昔洛韦3.1g,纯度只有93.95%,达不到分析检测使用的要求。
[0022]
本发明与方法3均以烯丙醇、多聚甲醛为起始原料,合成异更昔洛韦的中间体乙酰氧甲氧基烯丙醚,方法3以烯丙醇二氯甲烷溶液中加入多聚甲醛通氯化氢生成,反应后经分水、干燥、浓缩后得到氯甲基烯丙醚,该反应以浓硫酸滴加入浓盐酸中产生氯化氢气体,氯化氢气体难以计量,氯化氢易与碳链双键加成,存在反应不完全、副产物较多的问题;以丙酮作溶剂,氯甲基烯丙醚与乙酸钾取代反应,反应结束,过滤浓缩得到乙酰氧甲氧基烯丙醚。同方法3较复杂操作相比,本发明操作简便,以烯丙醇、多聚甲醛为起始原料,少量浓硫酸作催化剂,合成的羟基甲氧基烯丙醚不需后处理,与酸酐起酰化反应后常压蒸馏得到乙酰氧甲氧基烯丙醚。同方法3较复杂操作相比,本发明操作简便,得到的乙酰氧甲氧基烯丙醚收率高,纯度也较高。
技术实现要素:
[0023]
因此,为克服现有技术中制备工艺复杂,不能批量生产的问题,本发明提供了一种异更昔洛韦的制备方法。
[0024]
本发明涉及一种异更昔洛韦的制备方法,包括以下步骤,以烯丙醇为原料经羟甲基醚化、酰化合成侧链化合物,侧链化合物与二乙酰鸟嘌呤发生缩合反应后,再经氧化水解即可得到异更昔洛韦。
[0025]
具体步骤为:
[0026]
步骤(1):生成侧链化合物乙酰氧甲氧基烯丙醚或丙酰氧甲氧基烯丙醚
[0027]
以烯丙醇为原料,于65~70℃经多聚甲醛和酸催化剂反应得到羟基甲氧基烯丙醚,羟基甲氧基烯丙醚与酸酐酯化反应得到侧链化合物乙酰氧甲氧基烯丙醚或丙酰氧甲氧基烯丙醚;其中,酸催化剂为硫酸、三氟乙酸中的一种或多种,酸酐为乙酸酐、丙酸酐的一种或多种;
[0028][0029]
r为ch
3-,ch3ch
2-[0030]
步骤(2):缩合反应
[0031]
侧链化合物乙酰氧甲氧基烯丙醚或丙酰氧甲氧基烯丙醚中加入有机溶剂,在催化剂存在下,与二乙酰鸟嘌呤发生缩合反应,制备得到n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤;其中,有机溶剂为dmf、甲苯中的一种或多种;催化剂为对甲苯磺酸、双(对硝基苯基)磷酸酯、对乙酰氨基苯磺酸中的一种或多种;
[0032][0033]
r为ch
3-,ch3ch
2-[0034]
步骤(3):氧化水解反应
[0035]
化合物n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤被高锰酸钾稀碱液氧化并在稀碱液下水解得到化合物9-(2,3-二羟基-1-丙氧甲基)鸟嘌呤,即产物异更昔洛韦;其中,碱为氢氧化钠、氢氧化钾中的一种或多种;
[0036][0037]
步骤(4):异更昔洛韦的精制
[0038]
将异更昔洛韦粗品用有机溶剂的水溶液精制,得到较高纯度的异更昔洛韦,可达98.9%以上,有机溶剂为甲醇、乙醇、异丙醇中的一种或多种。
[0039]
本发明所采用的制备工艺路线简单易操作,得到的异更昔洛韦纯度较高,纯度可达98.90%以上,不需硅胶柱层析,能批量合成,实验室可合成几十克以上的异更昔洛韦。与
现有公开的异更昔洛韦制备工艺路线相比,具有工艺路线短、操作简单、批量得到产物的优点。本发明的有益效果是:具有工艺路线短、原料易得、操作简单、纯度高、易于批量合成的特点。
具体实施方式
[0040]
现在结合具体实施例对本发明作进一步说明,以下实施例旨在说明本发明而不是对本发明的进一步限定。
[0041]
实施例一:乙酰氧甲氧基烯丙醚的制备
[0042]
100g烯丙醇中加入多聚甲醛后82.4g,浓硫酸0.8g,于65~70℃保温反应6h。反应毕,冷却至0~10℃,反应液中滴加乙酸酐347.3g,滴加毕在低温下搅拌1h后,逐渐升温至30~35摄氏度保温10~12h,保温毕,于常压蒸馏144~150℃接收乙酰氧甲氧基烯丙醚馏份160.1g以上,气相纯度92%以上。
[0043]
实施例二:乙酰氧甲氧基烯丙醚的制备
[0044]
100g烯丙醇中加入多聚甲醛后82.4g,三氟乙酸1.2g,于65~70℃保温反应6h。反应毕,冷却至0~10℃,反应液中滴加乙酸酐347.3g,滴加毕在低温下搅拌1h后,逐渐升温至30~35摄氏度保温10~12h,保温毕。于常压蒸馏144~150℃接收乙酰氧甲氧基烯丙醚馏份148.3g以上,气相纯度92%以上。
[0045]
实施例三:丙酰氧甲氧基烯丙醚的制备
[0046]
100g烯丙醇中加入多聚甲醛后82.4g,浓硫酸0.8g,于65~70℃反应6h。反应毕,冷却至0~10℃,反应液中滴加丙酸酐469.7g,加毕在低温下搅拌1h后,逐渐升温至30~35摄氏度保温10~12h。保温毕,于常压蒸馏149~156℃接收丙酰氧甲氧基烯丙醚馏份174.8g以上,纯度92%以上。
[0047]
实施例四:丙酰氧甲氧基烯丙醚的制备
[0048]
100g烯丙醇中加入多聚甲醛后82.4g,三氟乙酸1.2g,于65~70℃反应6h。反应毕,冷却至0~10℃,反应液中滴加丙酸酐496.7g,加毕在低温下搅拌1h后,逐渐升温至30~35摄氏度保温10~12h。保温毕,于常压蒸馏149~156℃接收丙酰氧甲氧基烯丙醚馏份157.6g以上,纯度92%以上。
[0049]
实施例五:n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤的制备
[0050]
160.0g乙酰氧甲氧基烯丙醚中加入640gdmf,加入72.1g二乙酰鸟嘌呤,加入对甲苯磺酸2.0g,于90~95℃保温20h。保温结束,蒸除溶剂,加入236g水,升温至回流后冷却降温至0~5℃,过滤,滤饼加少量冷水淋洗,烘干得n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤81.7g以上,纯度96.8%以上。
[0051]
实施例六:n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤的制备
[0052]
160.0g乙酰氧甲氧基烯丙醚中加入800g甲苯,加入72.1g二乙酰鸟嘌呤,加入对甲苯磺酸2.0g,于90~95℃保温25h。保温结束,蒸除溶剂,加入236g水,升温至回流后冷却降温至0~5℃,过滤,滤饼加少量冷水淋洗,烘干得n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤75.3g以上,纯度96.8%以上。
[0053]
实施例七:n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤的制备
[0054]
160.0g乙酰氧甲氧基烯丙醚中加入640.0gdmf,加入72.1g二乙酰鸟嘌呤,加入对
乙酰氨基苯磺酸2.0g,于90~95℃保温22h。保温结束,蒸除溶剂,加入236g水,升温至回流后冷却降温至0~5℃,过滤,滤饼加少量冷水淋洗,烘干得n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤78.9g以上,纯度96.8%以上。
[0055]
实施例八:n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤的制备
[0056]
160.0g乙酰氧甲氧基烯丙醚中加入800.0g甲苯,加入72.1g二乙酰鸟嘌呤,加入对乙酰氨基苯磺酸2.0g,于90~95℃保温28h。保温结束,蒸除溶剂,加入236g水,升温至回流后冷却降温至0~5℃,过滤,滤饼加少量冷水淋洗,烘干得n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤76.3g以上,纯度96.8%以上。
[0057]
实施例九:n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤的制备
[0058]
160.0g乙酰氧甲氧基烯丙醚中加入640gdmf,加入72.1g二乙酰鸟嘌呤,加入双(对硝基苯基)磷酸酯2.0g,于90~95℃保温。保温结束,蒸除溶剂,加入236g水,升温至回流后冷却降温至0~5℃,过滤,滤饼加少量冷水淋洗,烘干得n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤79.2g以上,纯度96.8%以上。
[0059]
实施例十:n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤的制备
[0060]
160.0g乙酰氧甲氧基烯丙醚中加入800g甲苯,加入72.1g二乙酰鸟嘌呤,加入双(对硝基苯基)磷酸酯2.0g,于90~95℃保温30h,保温结束,蒸除溶剂,加入236g水,升温至回流后冷却降温至0~5℃,过滤,滤饼加少量冷水淋洗,烘干得n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤75.4g以上,纯度96.8%以上。
[0061]
实施例十一:n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤的制备
[0062]
170.0g丙酰氧甲氧基烯丙醚中加入600gdmf,加入67.1g二乙酰鸟嘌呤,加入对甲苯磺酸2.0g,于90~95℃保温23h。保温结束,蒸除溶剂,加入221g水,升温至回流后冷却降温至0~5℃,过滤,滤饼加少量冷水淋洗,烘干得n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤70.1g以上,纯度96.8%以上。
[0063]
实施例十二:n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤的制备
[0064]
170.0g丙酰氧甲氧基烯丙醚中加入750g甲苯,加入67.1g二乙酰鸟嘌呤,加入对甲苯磺酸2.0g,于90~95℃保温27h。保温结束,蒸除溶剂,加入221g水,升温至回流后冷却降温至0~5℃,过滤,滤饼加少量冷水淋洗,烘干得n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤65.3g以上,纯度96.8%以上。
[0065]
实施例十三:n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤的制备
[0066]
170.0g丙酰氧甲氧基烯丙醚中加入600.0gdmf,加入67.1g二乙酰鸟嘌呤,加入对乙酰氨基苯磺酸2.0g,于90~95℃保温24h,保温结束,蒸除溶剂,加入221g水,升温至回流后冷却降温至0~5℃,过滤,滤饼加少量冷水淋洗,烘干得n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤63.7g以上,纯度96.8%以上。
[0067]
实施例十四:n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤的制备
[0068]
170.0g丙酰氧甲氧基烯丙醚中加入750.0g甲苯,加入67.1g二乙酰鸟嘌呤,加入对乙酰氨基苯磺酸2.0g,于90~95℃保温29h。保温结束,蒸除溶剂,加入221g水,升温至回流后冷却降温至0~5℃,过滤,滤饼加少量冷水淋洗,烘干得n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤62.5g以上,纯度96.8%以上。
[0069]
实施例十五:n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤的制备
[0070]
170.0g丙酰氧甲氧基烯丙醚中加入600gdmf,加入67.1g二乙酰鸟嘌呤,加入双(对硝基苯基)磷酸酯2.0g,于90~95℃保温26h。保温结束,蒸除溶剂,加入221g水,升温至回流后冷却降温至0~5℃,过滤,滤饼加少量冷水淋洗,烘干得n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤59.7g以上,纯度96.8%以上。
[0071]
实施例十六:n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤的制备
[0072]
170.0g丙酰氧甲氧基烯丙醚中加入750g甲苯,加入67.1g二乙酰鸟嘌呤,加入双(对硝基苯基)磷酸酯2.0g,于90~95℃保温32h。保温结束,蒸除溶剂,加入221g水,升温至回流后冷却降温至0~5℃,过滤,滤饼加少量冷水淋洗,烘干得n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤57.4g以上,纯度96.8%以上。
[0073]
实验例十七:9-(2,3-二羟基-1-丙氧甲基)鸟嘌呤(即异更昔洛韦)的制备
[0074]
2%浓度的naoh水溶液320g中加入52.0gn
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤,31.4g高锰酸钾溶于410g 1%浓度的naoh水溶液,配制高锰酸钾稀碱溶液。n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤碱水溶液冷却至0~5℃,于0~5℃滴加高锰酸钾稀碱液。高锰酸钾稀碱液滴加毕,于0~5℃保温1~2h,保温毕,升温至室温20~30℃,于室温搅拌5~8h,搅拌毕,过滤,滤液冷却至10℃以下,加入10%盐酸水溶液中和至ph值为7,过滤,滤饼烘干得异更昔洛韦粗品35.6g以上;
[0075]
实验例十八:9-(2,3-二羟基-1-丙氧甲基)鸟嘌呤(即异更昔洛韦)的制备
[0076]
2%浓度的koh水溶液320g中加入52.0gn
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤,31.4g高锰酸钾溶于410g 1%浓度的naoh水溶液,配制高锰酸钾稀碱溶液。n
2-乙酰基-9-[甲氧基烯丙醚]鸟嘌呤碱水溶液冷却至0~5℃,于0~5℃滴加高锰酸钾稀碱液。高锰酸钾稀碱液滴加毕,于0~5℃保温1~2h,保温毕,升温至室温20~30℃,于室温搅拌5~8h,搅拌毕,过滤,滤液冷却至10℃以下,加入10%盐酸水溶液中和至ph值为7,过滤,滤饼烘干得异更昔洛韦粗品35.2g以上;
[0077]
实验例十九:异更昔洛韦的精制
[0078]
35.0g异更昔洛韦粗品加入到1300g 60%乙醇水溶液中,升温至回流,溶清,搅拌10min,冷却至0~5℃,于0~5℃搅拌1h,过滤,滤饼烘干得化合物5干品19.2g以上,为最终产品异更昔洛韦,纯度98.90%以上。
[0079]
实验例二十:异更昔洛韦的精制
[0080]
35.0g异更昔洛韦粗品加入到1350g 60%甲醇水溶液中,升温至回流,溶清,搅拌10min,冷却至0~5℃,于0~5℃搅拌1h,过滤,滤饼烘干得异更昔洛韦干品19.2g以上,纯度98.90%以上。
[0081]
实验例二十一:异更昔洛韦的精制
[0082]
35.0g异更昔洛韦粗品加入到1430g 65%异丙醇水溶液中,升温至回流,溶清,搅拌10min,冷却至0~5℃,于0~5℃搅拌1h,过滤,滤饼烘干得异更昔洛韦干品19.2g以上,纯度98.90%以上。
[0083]
本发明所采用的制备工艺路线简单易操作,得到的异更昔洛韦纯度较高,纯度可达98.90%以上,不需硅胶柱层析,能批量合成,实验室可合成几十克以上的异更昔洛韦。与现有公开的异更昔洛韦制备工艺路线相比,具有工艺路线短、操作简单、批量得到产物的优点。本发明的有益效果是:具有工艺路线短、原料易得、操作简单、纯度高、易于批量合成的
特点。
[0084]
以上述依据本发明的理想实施例为启示,通过上述的说明内容,相关工作人员完全可以在不偏离本项发明技术思想的范围内,进行多样的变更以及修改。本项发明的技术性范围并不局限于说明书上的内容,必须要根据权利要求范围来确定其技术性范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1