全基因组且靶向的单体型重构的制作方法
本申请是基于申请日为2014年7月18日,优先权日为2013年7月19日,申请号为201480051354.7,发明名称为:“全基因组且靶向的单体型重构”的专利申请的分案申请。
对相关申请的交叉引用
本申请要求2013年7月19日提交的美国临时申请号61/856,486和2013年9月4日提交的美国临时申请号61/873,671的优先权。所述申请的内容通过提述以其整体并入本文。
发明领域
本发明涉及用于单体型确定,且特别是在全基因组水平的单体型确定,以及靶向单体型确定的方法。
发明背景
dna鸟枪法测序技术的快速进步使得能够系统性鉴定个体的遗传变体(wheeler等,nature452,872-876(2008);pushkarev等,naturebiotechnology27,847-850(2009);kitzman等,sciencetranslationalmedicine4,137ra176(2012);和levy等,plosbiology5,e254(2007))。然而,由于人类基因组由同源的两组染色体组成,了解个体真正的遗传组成要求描绘遗传材料的母本和父本拷贝,或单体型(haplotype)。在个体中获得单体型的效用可以有几重:首先,单体型在临床上对于器官移植中供体-受体匹配结果的预测有用(crawford等,annualreviewofmedicine56,303-320(2005)和petersdorf等,plosmedicine4,e8(2007))并越来越多地用作检测疾病相关性的方法(studies等,nature447,655-660(2007);cirulli等,naturereviews.genetics11,415-425(2010);和ng等,naturegenetics42,30-35(2010))。第二,在表现出复合杂合性(compoundheterozygosity)的基因中,单体型提供了关于两种有害变体是否定位于相同的或不同的等位基因上的信息,这极大地影响了这些变体的遗传是否有害的预测(musone等,naturegenetics40,1062-1064(2008);和erythematosus等,naturegenetics40,204-210(2008);和zschocke,journalofinheritedmetabolicdisease31,599-618(2008))。在复杂的基因组(例如人)中,复合杂合性可能涉及在非编码顺式调节位点处的遗传或表观遗传变异,所述非编码顺式调节位点定位于远离它们调节的基因(sanyal等,nature489,109-113(2012)),这强调了获得染色体跨度(chromosome-span)的单体型的重要性。第三,来自个体组的单体型提供了关于群体结构的信息(internationalhapmap,c.等,nature449,851-861(2007);genomesproject,c.等,nature467,1061-1073(2010);和genomesproject,c.等,nature491,56-65(2012)),以及人种的进化史(meyer等,science338,222-226(2012))。最后,最近描述的基因表达中普遍的等位基因失衡(allelicimbalances)提示等位基因之间的遗传或表观遗传差异可能促成表达的定量差异(gimelbrant等,science318,1136-1140(2007);kong等,nature462,868-874(2009);xie等,cell148,816-831(2012);和mcdaniell等,science328,235-239(2010))。因此,对单体型结构的理解对于描绘促成这些等位基因失衡的变体机制将是关键的。总的来说,个体中完全单体型结构的知识对于推进个体化医疗至关重要。
认识到单体型的重要性,几个小组已经寻求扩展在群体和个体水平两者上对单体型结构的理解。诸如internationalhapmap工程和1000基因组工程等倡导已经尝试着通过连锁不平衡测量(linkagedisequilibriummeasures)基于无关个体群体测序数据或通过家庭三人组(familytrios)的基因型分析(genotyping)系统性重构(reconstruct)单体型。然而,使用该方法产生的准确定相(phased)的单体型的平均长度被限制为约300kb(fan等,naturebiotechnology29,51-57(2011)和browning等,americanjournalofhumangenetics81,1084-1097(2007))。也开发了许多实验方法来促进对个体的单体型定相,包括lfr测序,配偶配对(mate-pair)测序,fosmid测序,以及基于稀释的测序(levy等,plosbiology5,e254(2007);bansal等,bioinformatics24,i153-159(2008);kitzman等,naturebiotechnology29,59-63(2011);suk等,genomeresearch21,1672-1685(2011);duitama等,nucleicacidsresearch40,2041-2053(2012);和kaper等,procnatlacadsciusa110,5552-5557(2013))。这些方法最多可以重构范围在几千碱基到约百万碱基的单体型,但没有一个能够达到染色体跨度的单体型。使用基于荧光辅助细胞分选(facs)的测序,染色体分离及随后的测序,以及基于染色体微切割(micro-dissection)的测序完成了全染色体单体型定相(fan等,naturebiotechnology29,51-57(2011);yang等,proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica108,12-17(2011);和ma等,naturemethods7,299-301(2010))。然而,这些方法是低分辨率的,因为它们只能对个体中一部分的杂合变体进行定相,更重要的是,它们的执行在技术上具有挑战性,或要求专业的仪器。最近,使用来自精子细胞的基因型分析,已经进行了全基因组单体型分析(kirkness等,genomeresearch23,826-832(2013))。虽然该方法可以以高分辨率产生基因组跨度的单体型,但它不适用于普通人群,并需要复杂的减数分裂重组模式的解卷积(deconvolution)。
与全基因组单体型分析一起,靶向单体型分析(targetedhaplotyping)也是重要的。特别地,对hla(人白细胞抗原)基因座的靶向单体型分析可以帮助用于器官移植的受体-供体匹配以及阐明顺式调节元件在基因活性中的作用。
计算分析已经表明,来自先前建立的dna鸟枪法测序方法的单体型重构中的重要因素是经测序的基因组片段的长度(tewhey等,naturereviews.genetics12,215-223(2011))。例如,与常规的基因组测序相比(片段或插入物大小约500bp),通过配偶对测序(matepairsequencing)可以获得较长的单体型(片段或插入物大小约5kb)。然而,对于这些片段可以多长存在技术限制。例如,克隆比使用fosmid克隆获得的dna片段更长的dna片段是困难的。因此,使用现有的鸟枪测序方法,难以产生超过1百万个碱基的单体型区块(block),即使是在超深度测序覆盖。
因此,对于在全基因组水平重构单体型的方法,以及靶向单体型分析的方法存在需要。
发明概述
本发明通过提供在全基因组水平重构单体型的方法以及在基因组的靶向区域重构单体型的方法,解决了上述未满足的需求。
因而,本发明的特征在于用于对生物体全染色体单体型分析的方法。所述方法包括提供所述生物体的细胞,其含有具有基因组dna的染色体组(asetofchromosomes);将所述细胞或其核与固定试剂(fixationagent)孵育一段时间并使用限制酶限制固定的dna,从而允许基因组dna的原位临近连接(proximity-ligationofthegenomicdnainsitu),从而形成连接的基因组dna;使所述连接的基因组dna片段化(fragmenting)以形成具有第一基因组dna片段和第二基因组dna片段的临近连接的复合物;获得多个临近连接的dna片段以形成文库;对所述多个临近连接的dna片段测序以获得多个序列读出,并组装所述多个序列读出以构建一个或多个染色体的染色体跨度的单体型。
本发明还提供用于对生物体的靶向单体型分析的方法。所述方法包括提供所述生物体的细胞,其含有具有基因组dna的染色体组;将所述细胞或其核与固定试剂孵育一段时间并使用限制酶限制固定的dna,以允许基因组dna的原位临近连接从而形成连接的基因组dna;使所述连接的基因组dna片段化以形成具有第一基因组dna片段和第二基因组dna片段的临近连接的复合物;使所述邻接连接的dna片段与一个或多个寡核苷酸接触,所述寡核苷酸与所述临近连接的片段的子集的预选择区域杂交,以提供与所述寡核苷酸杂交的临近连接的片段的子集,将所述临近连接的片段的子集与所述寡核苷酸分离;对所述临近连接的dna片段的子集测序以获得多个序列读出,并组装所述多个序列读出以构建靶向单体型。在一个实施方案中,所述寡核苷酸是固定化的。
在某些实施方案中,所述方法进一步包括在孵育步骤之前从所述细胞分离细胞核。分离细胞核的方法是本领域已知的。例如,用于从植物细胞分离核的方法由lee等,(2007)theplantcell19:731-749公开。
在一些实施方案中,所述方法进一步包括在片段化步骤之前纯化连接的基因组dna。在其它实施方案中,所述方法进一步包括,在片段化步骤之后,使用标志物标记所述第一基因组dna片段或所述第二基因组dna片段;将所述第一基因组dna片段和所述第二基因组dna片段连接,使得所述标志物位于其间以形成标记的嵌合dna分子;以及剪切标记的嵌合dna分子以形成标记的、临近连接的dna片段。
在上述方法中,可以通过本领域已知的多种方法进行所述片段化步骤。例如,其可以通过酶促剪切进行,包括由限制酶,dna酶,或转座酶介导的。在一个实施方案中,通过使用限制酶消化连接的基因组dna以形成消化的基因组dna片段来进行该步骤。可以使用任意适合的限制酶(例如,bamhi,ecori,hindiii,ncoi,或xhoi)或两种或更多这些限制酶的组合。所述固定试剂可以包含甲醛,戊二醛,或福尔马林。可以通过使用标记有标志物(例如生物素)的核苷酸填充所述第一或第二基因组dna片段的末端来进行所述标记步骤。在该情况下,可以使用链霉亲合素进行所述获得步骤,所述链霉亲合素可以粘附到珠。对于连接步骤,它可以通过使用连接酶连接所述第一基因组dna片段和第二基因组dna片段来进行。所述连接步骤可以在溶液中或固体基质上进行。在固体基质上连接在本文中称为“栓系染色体捕获(tetheredchromosomalcapture)”。对于测序,它可以使用配对末端测序(pair-endsequencing)进行。
在本发明的一个实施方案中,每个配对末端测序读出片段长度的长度可以是至少20bp,例如长度为20-1000bp或优选20-150bp(例如,长度为20,25,30,40,50,60,60,80,90,100,110,120,130,140,或150bp)。对于每个染色体的单体型分析,文库含有至少15x序列覆盖,例如25-20x序列覆盖。优选的是,所述第一基因组dna片段和第二基因组dna片段在同一条染色体上或为顺式。优选的是,所述第一基因组dna片段和第二基因组dna片段在原位相距至少100bp,例如100-100mb(例如,100bp,1kb,10kb,1mb,10mb,20mb,30mb,40mb,50mb,60mb,70mb,80mb,90mb,或100mb)。
所述方法可以用于多种生物体,包括原核生物和真核生物。所述生物体包括真菌,植物和动物。在一个优选的实施方案中,所述生物体是植物。在另一个优选的实施方案中,所述生物体是哺乳动物或哺乳动物胚胎,或人或人胚胎。在一个实施方案中,所述人是器官的供体或受体。在这种情况下,所述器官在移植到具有匹配单体型的受体之前,可以使用本发明的方法进行单体型分析。本发明的方法可以用于二倍体细胞,非整倍体细胞,或多倍体细胞,例如,某些癌性细胞。
本发明的一个或多个实施方案的详细说明列于以下说明书中。本发明的其它特征,目的以及优势将从说明书和权利要求变得显而易见。
本发明包括以下实施方案:
实施方案1.一种用于对生物体全染色体单体型分析的方法,包括:
提供所述生物体的细胞,其含有具有基因组dna的染色体组;
将所述细胞或其核与固定试剂孵育一段时间以允许所述基因组dna原位交联,并且从而形成交联的基因组dna;
片段化所述交联的基因组dna,并连接临近定位的交联并片段化的基因组dna以形成临近连接的复合物,所述复合物具有第一基因组dna片段和第二基因组dna片段;
剪切所述临近连接的复合物以形成临近连接的dna片段;
获得多个所述临近连接的dna片段以形成文库;
对所述多个临近连接的dna片段测序以获得多个序列读出,和
组装所述多个序列读出以构建一个或多个染色体的染色体跨度的单体型。
实施方案2.一种用于生物体的靶向单体型分析的方法,包括提供所述生物体的细胞,其含有具有基因组dna的染色体组;将所述细胞或其核与固定试剂孵育一段时间以允许所述基因组dna的原位交联,并且从而形成交联的基因组dna;片段化所述交联的基因组dna,并连接临近定位的交联并片段化的基因组dna以形成临近连接的复合物,所述复合物具有第一基因组dna片段和第二基因组dna片段;剪切所述临近连接的复合物以形成临近连接的dna片段;使所述临近连接的dna片段与一个或多个寡核苷酸接触,所述寡核苷酸与所述临近连接的片段的子集的预选择区域杂交,以提供与所述寡核苷酸杂交的临近连接的片段的子集,将所述临近连接的片段的子集与所述寡核苷酸分离;对所述临近连接的dna片段的子集测序以获得多个序列读出,并组装所述多个序列读出以构建靶向单体型。
实施方案3.实施方案2的方法,其中所述寡核苷酸固定化到固体基质上。
实施方案4.实施方案1或2的方法,进一步包括在孵育步骤前从所述细胞分离细胞核。
实施方案5.实施方案1或2的方法,进一步包括在片段化步骤前纯化连接的基因组dna。
实施方案6.实施方案1或2的方法,进一步包括在片段化步骤后,
使用标志物标记所述第一基因组dna片段或所述第二基因组dna片段;
连接所述第一基因组dna片段和所述第二基因组dna片段,使得所述标志物在它们之间以形成标记的嵌合dna分子;和
剪切所述标记的嵌合dna分子以形成标记的、临近连接的dna片段。
实施方案7.实施方案1或2的方法,其中通过使用限制酶消化所述连接的基因组dna以形成消化的基因组dna片段进行所述片段化步骤。
实施方案8.实施方案1或2的方法,其中所述固定试剂包括甲醛,戊二醛,或福尔马林。
实施方案9.实施方案6的方法,其中通过使用标记有所述标志物的核苷酸填充所述第一或第二基因组dna片段的末端进行所述标记步骤。
实施方案10.实施方案9的方法,其中所述标志物是生物素。
实施方案11.实施方案10的方法,其中使用链霉亲合素进行获得步骤。
实施方案12.实施方案11的方法,其中所述链霉亲合素固定到珠。
实施方案13.实施方案6的方法,其中通过使用连接酶连接所述第一基因组dna片段和所述第二基因组dna片段进行连接步骤。
实施方案14.实施方案13的方法,其中在溶液中进行连接。
实施方案15.实施方案13的方法,其中在固体基质上进行连接。
实施方案16.实施方案1或2的方法,其中使用配对末端测序片段的配对末端测序进行测序。
实施方案17.实施方案16的方法,其中每个配对末端测序读出片段的长度为至少20bp。
实施方案18.实施方案16的方法,其中每个配对末端测序读出片段的长度为20-150bp。
实施方案19.实施方案16的方法,其中每个配对末端测序读出片段的长度为20,25,30,40,50,60,70,80,90,100,110,120,130,140,或150bp。
实施方案20.实施方案1或2的方法,其中对于每条染色体,所述文库含有至少15x序列覆盖。
实施方案21.实施方案20的方法,其中对于每条染色体,所述文库含有至少25-30x序列覆盖。
实施方案22.实施方案18的方法,其中所述第一基因组dna片段和所述第二基因组dna片段在同一条染色体上。
实施方案23.实施方案22的方法,其中所述第一基因组dna片段和所述第二基因组dna片段原位相距至少100bp。
实施方案24.实施方案23的方法,其中所述第一基因组dna片段和所述第二基因组dna片段原位相距100bp–100mb。
实施方案25.实施方案24的方法,其中所述第一基因组dna片段和所述第二基因组dna片段原位相距100bp,1kb,10kb,1mb,10mb,20mb,30mb,40mb,50mb,60mb,70mb,80mb,90mb,或100mb。
实施方案26.实施方案1或2的方法,其中所述生物体是真核生物。
实施方案27.实施方案1或2的方法,其中所述生物体是真菌。
实施方案28.实施方案1或2的方法,其中所述生物体是植物。
实施方案29.实施方案1或2的方法,其中所述生物体是动物。
实施方案30.实施方案1或2的方法,其中所述生物体是哺乳动物或哺乳动物胚胎。
实施方案31.实施方案1或2的方法,其中所述生物体是人或人胚胎。
实施方案32.实施方案31的方法,其中所述人是器官的供体或受体。
实施方案33.实施方案32的方法,其中在将所述器官移植到具有匹配单体型的受体前对所述器官进行单体型分析。
实施方案34.实施方案1或2的方法,其中所述细胞是二倍体细胞。
实施方案35.实施方案1或2的方法,其中所述细胞是非整倍体细胞。
实施方案36.实施方案1或2的方法,其中所述细胞是癌性细胞。
附图简述
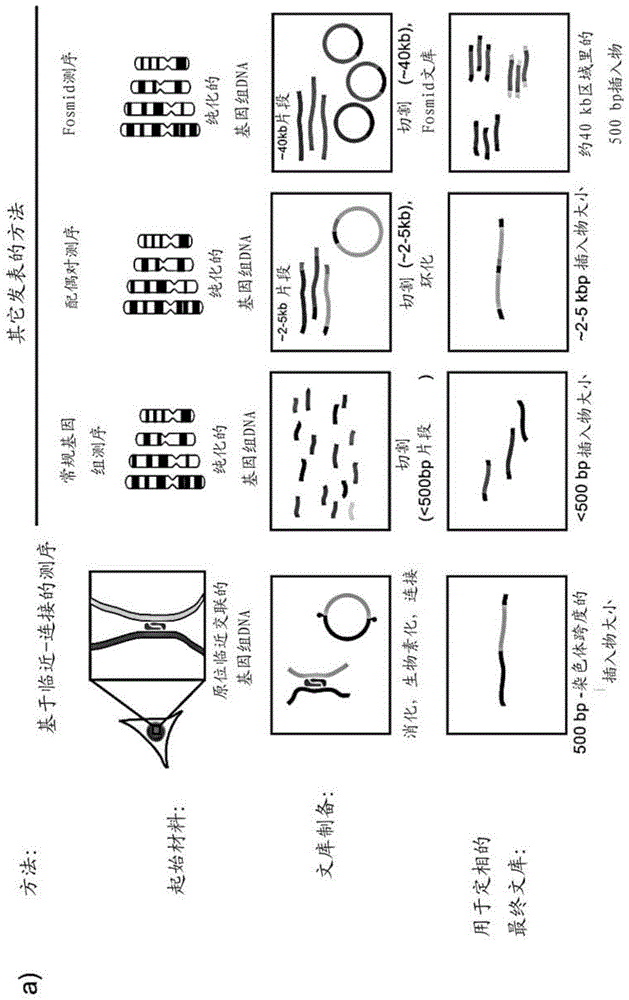
图1a-c是一组示出haploseq与其它用于重构生物体的单体型的方法比较的图:(a)图概述了几种用于对单体型定相的方法;(b)来自常规全基因组测序(wgs),配偶对和hi-c的插入物大小的频率分布;(c)图显示了临近连接读出在构建染色体跨度的单体型中的作用。
图2a-c是一组示出临近连接产物主要是单体型内(intra-haplotype)的图;(a)全基因组相互作用频率热图;(b)任意两个片段之间相互作用频率(log10比例)作为线性距离的函数;(c)比较h-反式相互作用(h-transinteraction)概率作为插入物大小的函数。
图3a-d是一组示出haploseq允许精确的、高分辨率、以及染色体跨度的单体型重构的图:(a)起源于129个等位基因的hi-c读出(上和下棒)的图,所述129个等位基因跨越第18号染色体的约30mb,并用于将变体连接到单个染色体跨度单体型中;(b)castxj129系统中基于hi-c的单体型定相的结果的表格;(c)通过模拟产生完全单体型的单体型定相方法的比较;(d)对单体型定相的经调整跨度(adjustedspan,as)的分析。
图4a-d是一组示出了使用haploseq在人gm12878细胞中单体型重构的图:(a)图证明了小鼠(castx129)和人(gm12878)之间在hoxd13/hoxd13基因上变体频率的差异;(b)表格描述了在castxj129系统中在低变体密度的情境下使用haploseq分析的单体型重构的完全性(“mvp区块中跨越的染色体%”),分辨率(“mvp区块中定相的变体%”),以及精确度(“mvp区块中定相的变体的精确度%”);(c)gm12878细胞中基于haploseq的单体型重构的结果的表格;(d)hi-c产生的种子单体型(seedhaplotype)跨越中着丝粒染色体的着丝粒。
图5a-d是一组示出了haploseq分析与局部条件定相(localconditionalphasing)结合允许在人中高分辨率单体型重构的图:(a)图描述了进行局部条件性定相的能力;(b)表格证明了gm12878细胞中局部条件性定相后使用haploseq的单体型定相的分辨率以及整体精确度;(c)图证明了在不同读出长度和覆盖的参数下,完成染色体跨度的种子单体型(seedhaplotype)(mvp区块)的能力;(d)点示出了读出长度和覆盖的不同组合产生高分辨率种子单体型的能力。
图6的图示出了每条castxj129染色体的h-反式相互作用的概率,其绘制为插入物大小的函数。
图7a-d是一组示出了单体型定相中完整性,精确度,以及分辨率的图形解释的图,(a)核苷酸碱基代表杂合snp,而“-”代表没有变异性(variability);(b)mvp区块的单体型定相表明分辨率;(c)先验已知的真实单体型和此知识帮助测量预测的从头单体型(de-novohaplotypes)的精确度,并且不精确的变体定相示于灰色方框位置;(d)不同的度量。
图8a-b是一组示出了限制的hapcut模型的图,所述模型仅允许到特定最大插入物大小(maxis)为止的片段,其中在较高的maxis,mvp区段的分辨率(a)较高但含有较高的精确度(b)。
图9是示出了捕获-hic实验方案的图表。
图10a-b示出了捕获-hic探针设计:(a)人类hla基因座(hg19)的ucsc基因组浏览器(genomebrowser)拍摄(shot)和(b)hla-dqb1基因放大的ucsc基因组浏览器拍摄,以证明探针靶向方法。
发明详述
高通量dna测序技术的快速进步加速了个体化医疗研究的步伐。虽然用于全基因组测序(wgs)数据组的变体发现和基因型分析的方法已经很好地建立了,但将染色体上的变体连接成单个的单体型仍然是一项挑战。
全基因组单体型分析和重构
本发明提供用于单体型分析的新方法,其包括临近连接和dna测序技术及用于单体型组装(haplotypeassembly)的概率算法(dekker等,science295,1306-1311(2002);lieberman-aiden等,science326,289-293(2009);kalhor等,naturebiotechnology30,90-98(2012);和bansal等,bioinformatics24,i153-159(2008))。称为“haploseq”(表示“使用临近连接和测序的单体型分析(haplotypingusingproximity-ligationandsequencing)”)的方法通过利用临近连接和dna鸟枪法测序重构完全单体型或靶向单体型。如本文所公开的,已经在先验已知完全单体型的杂种小鼠胚胎干细胞系和人类类淋巴母细胞细胞系中实验证实了haploseq。本文证明了使用haploseq,可以在小鼠中完成染色体跨度的单体型的重构,以约99.5%的精确度连接超过95%的等位基因。在人细胞系中,使用仅17x覆盖的基因组测序,将haploseq与局部条件性定相结合,从而在约81%分辨率上以约98%精确度获得染色体跨度的单体型。这些结果建立了临近连接和测序用于人群中单体型分析的效用。
本发明的haploseq方法的一个实施方案示于图1中。简单的说,图1a描述了haploseq和其它用于重构个体单体型的方法的比较。该图概述了几种用于定相单体型的方法。与以前的方法不同,临近-连接联系空间上靠近的远端dna片段。接着将这些片段从细胞分离并测序。
图1b示出了来自常规wgs,配偶对(gnerre,s.等,proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica108,1513-1518(2011))和hi-c的插入物大小的频率分布。x轴为碱基对(log10比例)。图代表1-22号染色体间从对gm12878细胞先前的发表取得的数据点的随机子集。在fosmid的情况下(kidd等,nature453,56-64(2008)),示出了比对后推断的克隆的大小分布。hi-c插入物大小得自发明人的实验室所生成的文库。插入物和克隆的大小与重构较长单体型的能力相关。在这些方法中,仅基于临近-连接的hi-c产生了丰富的长片段。
图1c显示了临近-连接读出在建立染色体跨度单体型中的作用。顶部和底部的序列代表两个同源染色体的区域,其中“-”代表无变异性而核苷酸代表杂合snp。杂合snp和插入缺失(indel)可以用于区分所述同源染色体。可以从短的插入物测序读出(i)建立局部单体型区块(“区块1”和“区块2”),这与常规的wgs或配偶对测序中的发生相似。考虑到变体之间的距离,这些小的单体型区块相对于彼此仍然是未定相的。可以使就线性序列而言远离定位的区域原位紧密接近(ii)。这些联系将通过临近-连接保存。大的插入物大小的临近-连接测序读出帮助将较小的单体型区块合并为单个染色体跨度的单体型(iii)。
hi-c技术是本领域已知的,且相关的方案可以在us20130096009和lieberman-aiden等,science326,289-293(2009)中找到,其内容通过提述并入本文。在一个实施方案中,所述hi-c方法包括纯化连接产物,接着进行大规模平行测序。在一个实施方案中,hi-c方法允许对跨越整个基因组的染色质相互作用的无偏爱(unbiased)鉴定。在一个实施方案中,所述方法可以包括以下步骤,其包括但不限于使用甲醛交联细胞;使用限制酶消化dna,留下5’-突出端(5’-overhang);填充所述5’-突出端,其包括生物素化的残基;以及在有利于交联的dna片段之间的连接事件的稀释条件下连接平端片段。在一个实施方案中,所述方法可以产生含有连接产物的dna样品,所述连接产物由核中最初紧密空间靠近的片段组成,在接合处使用生物素残基标记。在一个实施方案中,所述方法进一步包括创建文库(即例如,hi-c文库)。在一个实施方案中,通过剪切dna和使用链霉亲合素珠子选择含有生物素的片段来创建所述文库。在一个实施方案中,接着,使用大规模平行dna测序分析所述文库,产生相互作用片段的目录(catalog)。见图1a。
如本文所公开以及图2所示,通过本发明的方法获得的临近-连接产物主要是单体型内(intra-haplotype)。为了该目的,图2a示出了全基因组相互作用频率的热图。基于亲本菌株已知的单体型结构,区分起源于cast(“c”)或j129(“j”)基因组的hi-c读出。使用10mb二进制大小(binsize)计算每条染色体的每个等位基因之间的相互作用频率。每条染色体的cast或j129等位基因主要以顺式相互作用,确认了个体等位基因发生在hi-c数据中看到的染色体领域(chromosometerritories)。插图示出了12到16号染色体的cast和j129等位基因的放大视图。此外,图2b示出了任意两个片段间相互作用频率(log10比例)作为线性距离的函数。根据先前的单体型信息,读出对(read-pair)区分为顺式(顶部)和h-反式(底部)相互作用。顺式的相互作用频率可以比h-反式更常见几个数量级。值得注意的是,在大的基因组距离上(>100mbp),顺式的相互作用频率接近h-反式的相互作用频率,且观察到<2%的总体h-反式相互作用。使用来自castxj129系统中1-19号染色体的数据生成了图。最后,图2c示出了作为插入物大小的函数的h-反式相互作用概率的比较。使用来自castxj129系统中的1-19号染色体的数据生成了图。以2%平滑(smoothing)进行lowess拟合。在30mb以下,读出是h-反式相互作用的概率≤5%(虚线)。因此,这一截留值用作最大插入物大小用于进一步分析。
本发明的haploseq方法允许精确的,高分辨率,以及染色体跨度的单体型重构。图3a示出了自129个等位基因产生的hi-c读出的图,所述129个等位基因跨越18号染色体的总共约30mb,并用于将变体连接为单个的染色体跨度的单体型。以黑色文本示出了hi-c读出的序列,其中变体位置以红色和下划线示出。参考基因组的序列为灰色。在变体位置以及基于hi-c数据预测的单体型处,使用了每种基因型先验的cast和j129单体型。在这四个碱基处,在鉴定已知的单体型结构方面hi-c生成了完美的匹配。接着hapcut可以使用这些杂合变体作为节点和这些重叠读出作为边缘以形成图形结构。
图3b中的表格示出了castxj129系统中基于hi-c的单体型定相的结果。“染色体的可定相跨度”栏列出了可定相碱基的数量(第一个和最后一个杂合变体之间的碱基对差)。“mvp区块中跨越的变体”栏中列出了每条染色体通过mvp区块跨越的杂合变体的总数,其为完全性的替代测量,并用作估算分辨率的分母。“mvp模块中跨越的染色体%”栏中列出了由预测的单体型跨越的可定相碱基的百分比。“mvp区块中定相的变体%”栏中列出了mvp区块中跨越的变体间定相的所有杂合变体的百分比。在最后一栏中列出的是每个定相的杂合变体的精确度。对于每一条染色体,发明人生成了完全的(跨越>99.9%的碱基),高分辨率的(定相的>95%杂合变体),以及精确的(正确定相>99.5%的杂合变体)单体型。
图3c进一步示出了通过模拟产生完全单体型的单体型定相方法的比较。发明人以20x覆盖模拟了常规鸟枪法测序(平均数=400,sd=100),配偶对(平均数=4500,sd=200)和fosmid(平均数=35000,sd=2500)的75个碱基对配对末端测序数据(19号染色体)。尽管第一个读出随机置于基因组中,但是基于上述的正态分布参数选择第二个读出。发明人二次采样(sub-sampled)了castxj129数据以生成20xhi-c片段,其用于haploseq分析。y轴代表mvp区块的跨度作为19号染色体可定相跨度的函数。haploseq中mvp区块跨越了整个染色体,而其它方法中mvp区块仅跨越了染色体的一部分。发明人还将每种方法的20x测序覆盖与20x常规wgs数据组合,总共为40x覆盖以在较高的覆盖上比较方法。
图3d示出了对单体型定相的调整跨度(as)的分析。所述as定义为该区块中定相的杂合变体的跨度和分数的乘积。通过每个模块中定相的杂合变体的数量对单体型区块排序(x轴为排序的)并将整个染色体中累积的as表示于y轴上。在haploseq的情况中,单独mvp区块跨越了100%的染色体并含有90%的定相变体。在其它方法中,百分比定相随着发明人纳入非mvp模块而累积地增加。虚线表示通过如上文所讨论与wgs数据组合增加的40x覆盖。
本发明的haploseq方法还允许进行人类细胞(例如gm12878细胞)中的单体型重构。为此,图4a证明了小鼠(castx129)和人(gm12878)之间在hoxd13/hoxd13基因中变体频率的差异。还示出了这些基因座中hi-c读出覆盖(log10比例)。hi-c读出在高snp密度(小鼠)的情况下更可能含有变体(显示为“覆盖snp的读出”)。这继而允许这些变体更容易连接到mvp区块。在低变体密度的情况下(人类),情况不是这样,因此存在“缺口”,其中变体相对于mvp区块仍然未定相。
此外,图4b中的表格示出了在castxj129系统中在低变体密度的情况下使用haploseq分析的单体型重构的完全性(“mvp模块中跨越的染色体%”),分辨率(“mvp区块中定相的变体%”),以及精确度(“mvp区块中定相的变体的精确度%”)。在castx129基因组中对变体二次取样,以具有每1500个碱基的1个杂合变体,并如上文所述进行定相。发明人继续产生了完全(>99%染色体跨度)和精确(>99%精确度)的单体型。然而,在低变体密度的情况下,定相的变体的分辨率有减少(约32%)。数字四舍五入到三位小数。
另外,图4c中的表格总结了gm12878细胞中基于haploseq的单体型重构的结果。所述结果示出了完全性(“mvp模块中跨越的染色体%”)和分辨率(“mvp区块中定相的变体%”)。发明人能够生成染色体跨度的单体型(>99%),尽管分辨率较低(约22%)。在gm12878细胞中,在与castxj129系统中的约30x比较时,发明人产生了约17x覆盖。因此,发明人观察到了在与低密度castxj129(32%)相比时更低的分辨率(22%)。数字四舍五入到三位小数。
如图4d所示,本发明的方法允许产生种子单体型,其跨越中着丝粒染色体的着丝粒。示出了2号染色体着丝粒任一侧的两个区域。这两个hi-c产生的种子单体型任意命名为“a”和“b”。从三人组测序(triosequencing)获悉的gm12878个体的实际单体型示于下方,任意命名为“a”和“b”。hi-c产生的种子单体型在着丝粒的两端都匹配实际单体型。值得注意的是,实际单体型中的一些变体仍然是未定相的,因此促成了种子单体型中的“缺口”。另外,实际单体型不含所有的变体,因为三人组测序以低深度进行,因此种子单体型含有不在实际单体型中的一些定相变体(例如,见aak1区域中的第三个变体)。
haploseq分析可以与其它技术共同使用,例如局部条件定相以允许人类中的高分辨率单体型重构。图5a)示出了进行局部条件性定相的能力。x轴是通过模拟产生的染色体跨度的种子单体型分辨率。顶部图组示出了使用未校正的(上方)和邻域校正的定相(下方,窗口大小=3)两者的局部条件性定相的误差率。由于邻域校正,一些变体不能局部地推断。底部图组示出了由于邻域校正(neighborhood)仍然未定相的变体百分比,其作为分辨率的函数。所有的模拟在gm12878的1号染色体中完成。
图5b中的表格证明了gm12878细胞中,在局部条件性定相后使用haploseq的单体型定相的分辨率以及总体精确度。使用局部条件性定相,平均而言本发明人将分辨率从约22%增加到了约81%。该表格也描述了由于邻域校正(nc)所致的分辨率丢失,其平均仅约3%。发明人使用窗口大小为3个种子单体型定相变体来检测局部定相的性能。除提高的分辨率外,发明人也得到了精确的单体型,具有总体精确度约98%。此处的精确度反应了初始haplpseq分析的mvp区块的误差和来自局部条件性定相的误差。对于一些染色体,由于较低的覆盖,精确度较低(见下文表1)。
图5c中的图也证明了在不同读出长度和覆盖参数时完成染色体跨度的种子单体型(mvp区块)的能力。在所有情况下,使用约15x可用覆盖可以获得染色体跨度的种子单体型。所有模拟在gm12878的1号染色体中完成。相似地,图5d中的图示出了读出长度和覆盖的不同组合产生高分辨率种子单体型的能力。在这种情况下,较长的读出长度有助于hi-c生成的种子单体型的较高的分辨率。所有的模拟在gm12878的1号染色体中完成。
发明人在本文中描述了一种重构生物体的染色体跨度的单体型的新策略。与从鸟枪法测序读出重构完全单体型的其它单体型分析方法相比,本文公开的方法可以生成染色体跨度的单体型(fan等,naturebiotechnology29,51-57(2011);yang等,proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica108,12-17(2011);和ma等,naturemethods7,299-301(2010))。该方法最适合用于临床和实验室环境,因为haploseq需要的试剂和仪器容易得到。此外,所述方法比基于精子细胞的基因型分析的方法更灵敏(apt)(kirkness等,genomeresearch23,826-832(2013)),因为其可以从任意个体或细胞系的完整细胞产生全基因组单体型。因此,haploseq在个体化医疗中具有极好的效用。在个体中对单体型的确定鉴定新的单体型-疾病关联,其中一些已经在较小规模鉴定(he等,americanjournalofhumangenetics92,667-680(2013);zeng等,geneticepidemiology28,70-82(2005);和chapman等,humanheredity56,18-31(2003))。另外,对于理解基因表达中的等位基因偏好,完全单体型将至关重要,其将有助于群体中的遗传和表观遗传多态性及其在分子水平的表型结果(gimelbrant等,science318,1136-1140(2007);kong等,nature462,868-874(2009);和mcdaniell等,science328,235-239(2010))。此外,haploseq可以用于鉴定癌细胞中的遗传多态性,其导致对癌症治疗药物的抗性,或是对癌症治疗药物的抗性的标志物。最后,虽然在以下实施例中,所述方法以二倍体细胞示例,但是实验和计算的改进允许在具有更高倍性的细胞中的单体型重构,例如癌细胞。这可以有助于理解遗传改变的结果,其经常在肿瘤发生中看到。
先前,临近-连接用于研究染色体的空间构造(lieberman-aiden等,science326,289-293(2009)),而不是全基因组水平的单体型确定。如本文所公开的,它也是一种在研究个体的遗传构成中有价值的工具。如本文中表明,基于临近-连接的方法不仅可以告知哪个顺式调节元件与哪个靶基因物理相互作用,也能告知这些中的哪些等位基因在同一条染色体上相连。临近-连接数据也可以以和wgs同样的方式用于基因型分析。虽然远离限制酶切割位点的变体不太可能进行基因型分析,这是由于邻接-连接方法例如hi-c的偏爱,但是可以补充进行未基因型分析的变体的基于群体的归因(populationbasedimputation)(browning等,americanjournalofhumangenetics81,1084-1097(2007))以完成增加的基因型调用(call)。由于所有这点可以使用单次实验完成,haploseq可以用作全基因组分析的一般工具。
靶向单体型分析和重构
haploseq也可以用于对不同区域的靶向单体型分析。一旦进行了连接步骤,且得到了临近连接的片段的文库,定制设计的寡核苷酸(其可以固定化到固体表面上)引入到溶液中的文库。这些寡核苷酸“靶向”特定的临近-连接片段并与这些临近-连接片段杂交。分离与这类寡核苷酸杂交的临近-连接片段以提供新的文库。此文库现在含有临近连接的片段的子集,其能够被定制的寡核苷酸捕获。将这些片段测序并组装以产生定向的单体型。这一方法对于不同区域的定向单体型分析有用。例如,对hla区域(也称为人类主要组织相容性复合基因座或人类白细胞抗原基因座)(其为约3.5mb)的定向单体型分析可以通过该方法进行。这种对hla区域的定向单体型分析在器官移植中在预测供体-受体匹配的结果中有用。
图9中示出的是此靶向单体型分析的示意性例子。首先,将细胞交联并固定,从而捕获空间上临近的dna元件(顶部左边)。接着,使用例如hindiii消化细胞,并使用生物素化的核苷酸填充片段化的末端,接着如hi-c方案中所进行的那样再连接消化的末端(顶部中间)。对hi-c片段进行pcr扩增后,最终的hi-c文库由hi-c双标签组成,其可以被生物素化的rna探针靶向,所述探针已经经设计来捕获特异性hi-c片段(顶部右边)。接着,使用寡核苷酸捕获技术(oct),可以进行rna探针和hi-c文库的溶液杂交。这里,一些hi-c片段已经被两种rna探针靶向,而其它片段仅被一种靶向,并且所有的非靶向序列将不被rna探针结合(底部右边)。接着,使用链霉亲合素包被的珠子来结合生物素化的rna:dna双链体(底部中间),从而从hi-c文库中提取靶向hi-c片段,并创建捕获-hic文库。珠子结合的hi-c文库接着进行pcr扩增,纯化,并进行下一代测序(底部左边)。
在下文的实施例中,上述方法用于单体型分析人类hla区域,其为约3.5mb。图10中示出了本实施例中使用的捕获-hic探针设计。首先使用suredesign软件集(agilent)计算地产生探针序列。图10a中示出了人类中hla基因座(hg19)的ucsc基因组浏览器拍摄。图10b示出了hla-dqb1基因放大的ucsc基因组浏览器拍摄,以证明该探针靶向方法。在这种情况下,发明人靶向限制酶切割位点附近的+/-400bp,所述限制酶用于制备hi-c文库,在此情况中是hindiii(“靶向区域”追踪)。对于靶向区域,以4x铺盖密度(tilingdensity)设计探针,其目标在于使得靶向序列的每个核苷酸被高达4个探针序列覆盖。还应当注意的是,探针自身不与hindiii切割位点重叠(“hla探针”追踪)。它也选择为不靶向在靶定区域内的任意序列,其被repeatmasker称为含有重复序列(“错过区域”和“repeatmasker”追踪)。
本文所讨论的靶向单体型分析方法,例如,所述捕获-hic方法,提供了将整个hla基因座定相到单个单体型区块的机会,使得能够更好地在细胞以及器官移植方案中预测hla型匹配。几项研究已经揭示了大量疾病相关的非编码变体,其与特定的hla基因或等位基因关联(trowsdale等,annualreviewofgenomicsandhumangenetics14,301-323,(2013)和trowsdale,immunologyletters137,1-8,(2011))。因此,通过描绘hla的单个单体型结构,可以系统地去卷积遗传变异在hla关联的疾病和表型中的作用。
如本文所证明的,捕获-hic方法通常保留常规hi-c实验检测的染色质相互作用测量。因此,捕获-hic可以用作在特定基因座获得长距离相互作用的方法。例如,利用捕获-hic可以揭示基因组印记(imprinting)背后的单体型解决的长距离相互作用机制。虽然几个小组目前使用4c和5c技术来研究靶向染色质相互作用(simonis等,naturegenetics38,1348-1354,(2006),和dostie等,genomeresearch16,1299-1309,(2006)),捕获-hic提供了更灵活的方法学。具体地,4c被限制为使用单个视点(viewpoint)对相互作用分析,而5c被复杂的引物设计,有限的通量,以及仅分析连续的基因组区域所限制。或者,捕获-hic可以应用于在单次实验中检测成几千个视点的相互作用,并能够以无偏爱的方式检索区域和定制的3d相互作用频率。特别地,捕获-hic提供了经改编以捕获任意散布的基因组元件的能力,鉴于所述元件与限制酶切割位点相对临近,因此可以应用到一般情况。例如,通过将捕获-hic应用到基因组范围的启动子或其它基因组元件,可以以空前的分辨率和相对低的成本生成基因组范围的3d调控相互作用图谱。
最近已经证明了hi-c方案在从头组装基因组中有用((burton等,natbiotechnol31,1119-1125,(2013)和kaplan等,natbiotechnol31,1143-1147,(2013))。由于捕获-hic获得了高质量的染色质相互作用数据组,与hi-c相似,这一方法学可以用于生成人类或其它大型基因组的复杂区域的二倍体组装,例如t细胞受体beta(trcb)基因座(spicuglia等,seminarsinimmunology22,330-336,(2010))。此外,在群体规模中进行高度杂合hla基因座的二倍体组装可以允许检测到新的结构变体并使得能够精确的描绘人类迁移模式以及进行关联研究以发现个体化医疗用于多种疾病状态。类似的,最近hi-c也被用于宏基因组学研究来去卷积复杂的微生物组混合物中存在的物种(beitel等,peerj,doi:10.7287/peerj.preprints.260v1(2014)和burton等,species-leveldeconvolutionofmetagenomeassemblieswithhi-c-basedcontactprobabilitymaps.g3,doi:10.1534/g3.114.011825(2014))。随着捕获-hic的出现,可以捕获不同的基因座,其是提供信息且有分辨力的,足以基于捕获的hi-c片段描绘物种混合物。总的来说,本文公开的捕获-hic和其靶向定相的应用以及其它应用使得能够在个体化临床基因组学以及生物医药研究中启用新的途径。
术语“标志物”或“接合标志物”,如本文所使用的,指代任意化合物或化学部分,其能够被整合到核酸中并可以提供用于选择性纯化的基础。例如,标志物可以包括但不限于,标记的核苷酸接头,标记的和/或修饰的核苷酸,切口平移,引物接头,或有标签的接头。术语“标记的核苷酸接头”指代一类包含任何核酸序列的标志物,所述核酸序列包含掺入(例如连接)另一个核酸序列中的标记。例如,所述标记可以用来选择性纯化核酸序列(即例如,通过亲和色谱法)。这种标记可以包括但不限于生物素标记,组氨酸标记(即6his),或flag标记。
术语“标记的核苷酸”,“标记的碱基”,或“修饰的碱基”指代包含与标志物附着的任何核苷酸碱基的标志物,其中所述标志物包含对于配体具有独特亲和力的特异性部分。或者,结合伴侣可以具有对接合标志物的亲和力。在一些例子中,所述标志物包括但不限于生物素标记,组氨酸标记(即6his),或flag标记。例如,可以认为datp-生物素是标记的核苷酸。在一些例子中,片段化的核酸序列可以使用标记的核苷酸进行平端化(blunting),接着平端连接。
术语“标记”或“可检测标记”如本文所使用的,指代任意组合物,其可以通过光谱学,光化学,生物化学,免疫化学,电学,光学或化学手段检测。这样的标记包括用于用标记的链霉亲合素缀合物染色的生物素,磁珠(例如,dynabeadstm),荧光染料(例如,荧光素,德克萨斯红(texasred),罗丹明(rhodamine),绿色荧光蛋白等),放射性标记(例如,3h,125i,35s,14c,或32p),酶(例如,辣根过氧化物酶,碱性磷酸酶以及其它通常用于elisa中的酶),和热量标记,例如胶体金或有色玻璃或塑料(例如,聚苯乙烯,聚丙烯、胶乳等)珠。本发明中考虑的标记可以通过许多方法检测。例如,可以使用胶片或闪烁计数器检测放射性标记,可以使用光检测器以检测发射光来检测荧光标志物。通常可以通过向酶提供底物并检测通过所述酶作用于所述底物产生的反应产物,来检测酶促标记,并通过仅可视化有色标记来检测热量标记。
术语“片段”指代比衍生它的序列短的任何核酸序列。片段可以是任意大小的,范围从几百万碱基和/或几千碱基到仅几个碱基长。实验条件可以决定预期的片段大小,包括但不限于,限制酶消化,超声处理,酸孵育,碱孵育,微流化等等。
术语“染色体”,如本文所使用的,指代天然发生的核酸序列,其包含一系列称为基因的功能区域,其通常编码蛋白。其它的功能区域可能包括microrna或长的非编码rna,或其它调控元件。这些蛋白可能具有生物学功能或它们直接与相同或其它染色体相互作用(即,例如,调控染色体)。
术语“基因组区域”或“区域”指代任意确定长度的基因组和/或染色体。例如,基因组区域可以指代多于一条染色体之间的关联(即,例如,相互作用)。或者,基因组区域可以指代完全染色体或部分染色体。此外,基因组区域可以指代染色体上特定的核酸序列(即,例如,阅读框和/或调控基因)。
术语“片段化”指代任意过程或方法,通过所述过程或方法,化合物或组合物被分离为较小的单元。例如,所述分离可以包括,但不限于,酶促剪切(即,例如,转座酶介导的片段化,作用于核酸的限制酶或作用于蛋白的蛋白酶),碱水解,酸水解,或热诱导的热脱稳定化。
术语“热图”指代数据的任意图形表示,其中二维图中的变量采用的数值以颜色表示。热图已经广泛地用于表示许多可比较的样品中许多基因的表达水平(例如,不同状态的细胞,来自不同患者的样品),如从dna微阵列获得的。
术语“基因组”指代任意的染色体组和它们含有的基因。例如,基因组可以包括但不限于,真核细胞基因组和原核细胞基因组。
术语“固定”,“固定化”或“固定的”指代任意的方法或过程,其固定化任意和所有的细胞过程。因此,固定的细胞在固定的时候准确维持了细胞内组分之间的空间关系。许多化学物能够提供固定,包括但不限于,甲醛,福尔马林,或戊二醛。
术语“交联”指代两个化合物之间任意合适的化学关联,使得它们作为一个单元进一步处理。这种稳定性可以基于共价和/或非共价键合。例如,核酸和/或蛋白可以通过化学试剂交联(即,例如,固定剂),使得它们在常规的实验室方法(即,例如,提取,洗涤,离心等等)过程中维持它们的空间关系。
术语“联接”是两个核酸序列通过接合标志物的独特连接。这种连接可以通过以下过程产生,其包括但不限于,片段化,使用标记的核苷酸填充,以及平端连接。这种联接反映了两个基因组区域的临近,从而提供了功能性相互作用的证据。为了促进测序分析,可以选择性地纯化包括接合标志物的联接。
术语“连接的”如本文所使用,指代两个核酸之间的任意连接,其通常包括磷酸二酯键。所述连接通常在辅因子试剂和能源(即例如,三磷酸腺苷(atp))的存在下,通过催化酶的存在(即例如,连接酶)而促进。
术语“限制酶”指代任意蛋白,其在特定的碱基对序列处剪切核酸。
术语“选择性纯化”指代任意过程或方法,通过它们可以将特定的化合物和/或复合体从混合物或组合物中去除。例如,这种过程可能基于亲和色谱法,其中待去除的特定化合物与所述混合物或组合物的剩余物相比,对于所述色谱底物具有更高的亲和力。例如,通过使混合物通过包含链霉亲合素的色谱柱,可以从所述混合物中选择性纯化使用生物素标记的核酸,所述混合物包含没有使用生物素标记的核酸。
术语“纯化的”或“分离的”指代下述的核酸组合物,其已经经历处理(例如,分级(fractionation))以去除各种其它组分,且该组合物基本保持了其表达的生物学活性。在使用术语“基本纯化”的情况下,此名称将指代下述组合物,其中核酸形成所述组合物的主要组分,例如组成所述组合物的约50%,约60%,约70%,约80%,约90%,约95%或更多(即例如,重量/重量和/或重量/体积)。术语“纯化至同质”用于包括已经纯化至“表观同质”,使得存在单个核酸序列(即例如,基于sds-page或hplc分析)的组合物。纯化的组合物不意图表示可以保留一些微量杂质。术语“基本纯化”指代从其天然环境中取出,分离或分开,并且至少60%不含,优选75%不含,更优选90%不含与它们天然相关的组分的分子(核酸或氨基酸序列)。因此,“分离的多核苷酸”指代基本纯化的多核苷酸。
“核酸序列”或“核苷酸序列”指代寡核苷酸或多核苷酸,以及其片段或部分,并指代基因组或合成来源的dna或rna,其可以是单链或双链的,并表示有义或反义链。
术语“分离的核酸”指代任意下述的核酸分子,其已经从其天然状态取出(例如,从细胞取出,在优选的实施方案中,不含其它基因组核酸)。
术语核苷酸的“变体”指代与参照寡核苷酸的不同之处在于具有缺失,插入和取代的新的核苷酸序列。这些可以使用多种方法(例如,测序,杂交测定法等等)检测。“缺失”定义为下述的核苷酸或氨基酸序列中的变化,其中分别缺少一个或多个核苷酸或氨基酸残基。“插入”或“增加”是下述的核苷酸或氨基酸序列中的变化,其已经导致增加一个或多个核苷酸或氨基酸残基。“取代”源自一个或多个核苷酸或氨基酸分别被不同的核苷酸或氨基酸置换。
术语“同源性”或“同源”如本文所使用的,就核苷酸序列而言指代达到与其它核苷酸序列互补性的程度。可以有部分同源性或完全同源性(即同一性)。与核酸序列部分互补,即“基本同源”的核苷酸序列是至少部分抑制完全互补序列与该靶核酸序列杂交的序列。可以使用杂交测定(southern或northern印迹,溶液杂交等等)在低严格性条件下检查对完全互补序列与靶序列杂交的抑制。在低严格性条件下,基本同源的序列或探针将竞争并抑制完全同源序列与靶序列的结合(即,杂交)。
术语“癌症治疗药物”用于本文指代所有的化疗试剂,癌细胞可以对随时化疗剂获得化学抗性。例子包括jak/stat抑制剂,p13激酶抑制剂,mtor抑制剂,erbb抑制剂,拓扑异构酶抑制剂,等等。
实施例
实施例1一般方法和材料
本实施例描述了用于以下实施例2-9中的一般方法和材料。
细胞培养和实验方法
f1代小家鼠(musmusculuscastaneus)xs129/svjae(f123系)是来自edithheard实验室的赠品,且先前已经描述于gribnau等,genes&development17,759-773(2003)。在含有敲除血清替换物(knockoutserumreplacement)的小鼠es细胞培养基中培养这些细胞:dmem85%,15%敲除血清替换物(invitrogen),青霉素/链霉素,1x非必需氨基酸(gibco),1xglutamax,1000u/mllif(millipore),0.4mmβ-巯基乙醇。f123小鼠es细胞最初在0.1%明胶包被的平板上培养,所述平板具有丝裂霉素-c处理的小鼠胚胎成纤维细胞(millipore)。在收获前,细胞在0.1%明胶包被的不含饲养者(feeder)的平板上传代两次。gm12878细胞(coriell)在补充有15%胎牛血清和1x青霉素/链霉素的85%rpmi培养基中悬浮培养。
在悬液中(gm12878)或在胰蛋白酶处理后(f123小鼠ex细胞)收获细胞。甲醛固定和hi-c实验如先前描述于lieberman-aiden等,science326,289-293(2009)中那样进行。
基因型分析
从depristo等,naturegenetics43,491-498(2011)下载gm12878的变体调用和基因型,并且这些用于单体型重构。从1000基因组工程(genomesproject,c.等,nature467,1061-1073(2010))下载gm12878的定相信息。1000基因组工程对gm12878的定相利用了低覆盖测序,因此仅覆盖该个体基因组中~约65%的经基因型分析的杂合变体(depristo等,naturegenetics43,491-498(2011))。值得注意的是,“gm12878”是类淋巴母细胞系的名称,而“na12878”是衍生该细胞系的个体的标识符。在此处的整个实施例中,为了一致和清楚,使用gm12878。
为了生成杂种castxj129细胞的基因型调用,从公众可用的数据库下载了亲本基因组测序数据。对于小家鼠,从欧洲核苷酸档案(europeannucleotidearchive)下载基因组序列(登录号erp000042)。从序列读出档案(sequencereadarchive)(登录号srx037820)下载了s129/svjae基因组测序数据。使用novoalign(www.novocraft.com)并使用samtools(li等,bioinformatics25,2078-2079(2009))将读出与mm9基因组比对,并且将未定位的读出和pcr重复过滤掉。使用genomeanalysistoolkit(gatk)(mckennaetal.,genomeresearch20,1297-1303(2010))处理最终比对的数据组。特别地,进行了插入缺失再比对和变体再校准。gatkunifiedgenotyper用于产生snp和插入缺失调用。发明人过滤掉了不满足gatk质量过滤器或称为杂合变体的变体,因为基因组测序在纯合亲本近交系小鼠中进行。亲本中的基因型调用都用于确定顺式相对于h-反式的相互作用程度,以及用于获知先验的杂种castxj129细胞定相到单体型重构。
hi-c读出比对
对于hi-c读出比对,将hi-c读出与mm9(小鼠)或hg18(人)基因组比对。在每种情况下,掩饰小家鼠或s129/svjae(对于小鼠)或gm12878(对于人)中在基因组中基因型分析为snp的任何碱基。为了减少参考偏爱定位假象(referencebiasmappingartifacts),这些碱基被掩饰为“n”。使用novoalign将hi-c读出作为单末端读出重复比对。特别的,对于重复比对,首先将整个测序读出与小鼠或人的基因组比对。接着,将未定位的读出修整(trim)5个碱基对,并再次比对。重复这一过程,直到读出与基因组成功比对或者直到修整的读出长少于25个碱基对。重复比对对于hi-c数据有用,因为某些读出跨越临近-连接的接合,并且由于缺口和错配而不能成功与基因组比对。当修整将跨越连接接合的读出的部分去除时,重复修整的未定位的读出允许这些读出与基因组的成功比对。在作为单末端的读出的重复比对完成后,使用内部脚本将读出手动配对。去除未定位的和pcr重复读出。然后,比对的数据组最终经过gatk插入缺失再比对和变体再校准。
同源染色体之间相互作用频率的分析
当比对hi-c数据时,配对末端读出可以具有被定位到同一条染色体上(染色体内)或定位到不同染色体(染色体间)的两个末端。然而,hi-c数据的最初定位利用单倍体参考基因组且不区分单个测序读出定位到染色体的两个同源拷贝的哪一个。结果,最初定位为“染色体内”的读出对被分解为发生于同一条同源染色体上的读出(其真正为顺式)和定位于两个同源对之间的读出(其被定义为“h-反式”)。
为了确定相对于h-反式为顺式的读出的程度,首先区分个体读出定位到哪个等位基因。这是通过鉴定与基因组中变体位置重叠的读出,并接着确定所述变体位置处测序的碱基对应哪个等位基因来完成的。一旦获得了该信息,可以确定相对于h-反式而言区域以顺式相互作用的频率(见图2c和6)。
如通过染色体内和染色体间读出定义的可用覆盖
对于使用hapcut的定相,可以利用染色体内和染色体间读出两者。对于染色体间读出,可以将每个染色体间读出对考虑为两个单末端读出,因为这些读出的配对信息不可用于定相。相反,考虑所有的染色体内读出用于定相。单个读出含有多于一个变体的概率很小,特别是在人类中,其中变体的密度相对较低。这与仅配对的染色体内读出将具有大的插入物大小的实情组合,意味着有助于单体型定相的成功的绝大部分读出是染色体内读出。因此,“可用覆盖”定义为仅源自染色体内读出的基因组覆盖。
hi-c实验在castxj129中生成约22%的染色体间读出,而gm12878中约55%的读出是染色体间的。换句话说,castxj129中795m中的620m配对末端读出是有用的,具有可用覆盖为30x。在人类中,577m中的262m配对末端读出是有用的,导致可用覆盖为17x。因此,尽管测序的读出总数相对类似,但人类中具有较低的可用覆盖。以发明人的经验,hi-c实验中相对于染色体间为染色体内的所有读出的分数可以在实验间和细胞类型间变化。
使用hapcut的haploseq
使用hapcut算法来进行haploseq的计算方面,其细节先前描述于bansal等,bioinformatics24,i153-159(2008)。hapcut最初设计为作用于常规基因组测序(wgs)或配偶对数据。hapcut构建具有杂合变体作为节点以及节点之间由相同片段覆盖的边缘的图表。因此,仅具有至少两个杂合变体的片段对于单体型定相是有用的。hapcut使用分选方法将这种“单体型信息性”片段从协调分选的bam文件中提取,所述分选方法将每个潜在的单体型信息性读出储存在缓存中,直到看到其配偶。缓存大小经定制以允许hapcut处理大的插入物大小的临近-连接读出。
hapcut使用贪婪最大切割试探法(greedymax-cutheuristic)以鉴定用于在mec得分函数下具有最低得分的图中的每个连接组分的单体型解决办法。具体地,最初的hapcut算法使用o(n)重复来寻找最佳切割。由于hi-c数据产生具有单个大连接组分的染色体跨度的单体型,该默认方法花费几天的计算时间来定相castxj129基因组。为了减少计算时间,评估了减少重复数量对定相的精确度的影响。对于cast*129系统,观察到将重复数量增加到超过1000没有显著改善精确度。对于gm12878,允许高达100,000个重复。将该解决方案重复多次,且在castxj129中最多使用了21次重复和gm12878中101次。与小鼠的数据相比,考虑到较低的变体密度和减少的序列覆盖,gm12878细胞中的参数允许hapcut获取较高的精确度。
最大插入物大小分析
如先前所提到的,相对于h-反式而言hi-c读出为顺式的概率作为两个读出对之间距离的函数变化(图2c)。在较短的基因组距离,染色体内读出为h-反式的概率非常低。在大的距离(>30mbp),这一概率大大提高且理论上更可能将错误的hapcut连接引入相中。考虑到这一点,使用了castxj129数据中的1号,5号,10号,15号和19号染色体的hi-c数据,且重复单体型重构,这允许可变的最大插入物大小值。插入物大小比可允许的最大插入物大小更大的任意读出被排除。该分析使用低变体密度情况进行,对于此分析因为较低的密度最适合用于人类应用(图8a-b)。该步骤导致haploseq分析增加的精确度伴随分辨率的中度降低。
插入物大小依赖概率修正
hapcut算法的一个有用的特征是,为了计算潜在单体型的得分,其负责变体位点的基础质量得分。换句话说,如果在连接两个变体的测序读出中并且一个变体位点的基础质量较低,则此读出在产生其最终单体型调用中被hapcut给予相对较低的权重。因此,hapcut可以使用该信息来试着忽略来自产生错误的单体型连接的潜在测序误差。如先前所提到的,在hi-c数据中,误差也可能由于h-反式相互作用而产生,其比测序误差频繁得多,且表现出距离依赖性行为。因此,尝试基于两个读出之间的距离,解决相对于h-反式而言相互作用为顺式的可能性。castx129hi-c数据用于鉴定顺式或h-反式的读出。将插入物大小二进制化(binned)成50kb二进制中,并评估读出为h-反式的可能性(#h-反式/(#cis+#h-反式)。接着,以2%平滑使用局部回归(lowess)以预测任意给定插入物大小的h-反式概率。对于每个染色体内读出,将顺式概率(1-h-反式)乘以基础质量来得到该染色体内读出为同源反式相互作用的几率。作为结果,在鉴定单体型解决办法中,对于更可能是h-反式的读出,hapcut给出较低的权重。
增加h-反式相互作用概率中度增加haploseq精确度,而对分辨率没有任何影响。作为比较,30mb的19号染色体maxis具有1.1%的误差率(图8b)。在增加了h-反式概率后,误差率为0.9%(图4b),其中误差率定义为1-精确度。
局部条件性定相模拟
为了研究以不同的分辨率百分比进行局部定相的能力,进行了逐步分析。首先,以不同的分辨率生成种子单体型。接着,使用beagle(v4.0)(browning等,genetics194,459-471(2013))在种子单体型的指导下进行局部定相。最后,局部定相的精确度通过将其与来自1000基因组计划的先验已知的定相信息比较来检查。
为了以不同分辨率模拟种子单体型,首先模拟种子基因型。使用读出长度和覆盖的不同组合来获得不同分辨率的种子基因型。具体地,来自h1和h1衍生细胞的hi-c染色体内读出起始位点(未发表数据)用于生成给定读出长度和覆盖的读出对。这允许维持hi-c数据结构和模拟数据中观察到的插入物大小的分布。为了生成种子基因型,发明人构建了具有节点和边缘的图,其中所述节点表示gm12878(1号染色体)中的杂合变体,所述边缘对应覆盖多个变体的读出。该图基本上是基因型图,因为该定相还未知。因此,该图的整个点是基于分辨率和hi-c数据结构,提供变体的子集,其为种子基因型的一部分,或不是通过局部定相推断的缺口。以要求的读出长度和覆盖参数生成种子基因型,来达到特定的分辨率。这些种子基因型用于局部定相(图5a)以研究用于产生足够分辨率的种子单体型的最低要求(图5c-d)。这两种分析独立进行并在这两种情况中,生成种子基因型和下游分析重复10次来记录平均结果。
为了进行局部条件性定相,需要先验的单体型系统来检查局部条件性定相的精确度。因为来自三人组的先验单体型信息仅覆盖约65%的杂合变体,决定仅在所述三人组子集上进行局部定相模拟。特别地,条件为作为种子基因型或“缺口”的一部分的每个变体应当是1000基因组定相的三人组的一部分。使用三人组信息将种子基因型转化为种子单体型,同时使“缺口”变体保持未定相。接着,使用beagle将种子单体型上的局部定相条件用于推断缺口变体的定相。允许同源变体辅助beagle来从隐藏markov模型获得更好的预测。
为了对种子单体型未定相变体进行邻域校正,发明人收集了在种子单体型中定相的各在上游和下游的3个变体。接着检查在种子单体型中存在的定相和由beagle预测的定相之间是否存在100%相关性。这给出了beagle能够在该“局部区域”表现得多好的信心。如果存在100%匹配,认为所述变体是条件性定相的。如果不是100%匹配,在最终的单体型中忽略未定相的变体。尝试了其它窗口大小,例如5和10,并且没有发现精确度的改进。
人gm12878细胞中的局部条件性定相
发明人将haploseq分析和局部条件性定相偶联以增加gm12878细胞中的分辨率。如早前所描述的对gm12878(ref.44)和群体样品之间共同的基因型进行了局部条件性定相。另外,由于所述种子单体型不是100%准确的,发明人标记了与局部定相不一致的种子单体型定相的变体。使这些标记的变体“未定相”,因为它们可以是潜在的错误。因此,除了使用用于决定缺口变体是否需要局部定相的邻域校正外(如在模拟中),本发明人还使用了这一信息来标记种子单体型中可能是潜在错误的变体。这允许局部定相后精确度的小幅增加(见表1)。
总体haploseq精确度估算为局部定相后mvp区块中正确定相的杂合变体的分数(图5b和表1)。具体地,发明人仅使用在三人组中定相的变体来评估精确度。对于chrx中的局部定相,发明人使得雄性单倍体基因型为纯合。
gm12878细胞具有比castxj129低的变体密度,并且较低的覆盖增加了对预测模型的更多限制,当与低密度castx129情况中的0.8%相比,导致相对较高的haploseq误差率2%。25-30x的可用覆盖(如图5c-d中所示)可以帮助增加精确度,并潜在地覆盖种子单体型中更多的稀有变体。目前,约16%的变体没有局部定相,这是由于它们在群体中的缺乏。这些可以通过额外的hi-c数据或甚至常规基因组测序数据(其可以潜在地将缺口变体连接到种子mvp区块中的变体)定相。haploseq分析的一个重要方面是形成种子染色体跨度的单体型的能力,其不能从常规基因组测序或配偶对或fosmid产生。
fosmid模拟
为了模拟基于fosmid的测序(图4b和c),发明人模仿了作为配对末端测序的fosmid克隆,其具有接近40kb的插入物大小。发明人推理该方法较容易模拟,并仍然保持fosmid添加到单体型重构的数据结构。作为证据,如由其他组报道的,所述模拟在人类中产生大小高达1mb的单体型区块(kitzman等,naturebiotechnology29,59-63(2011);suk等,genomeresearch21,1672-1685(2011);和duitama等,nucleicacidsresearch40,2041-2053(2012))。
为了该目的,对于gm12878的1号染色体,模拟的100bp配对末端读出为各种测序覆盖。使用随机起始位点模拟读出,其具有提到的平均插入物大小和平均值的10%标准偏差。fosmid插入物表示使用“fosmid大小”的插入物的模拟,以精确定点(pinpoint)这些大型片段产生较长单体型的能力。500bp偏态混合(skewedmix)插入物含有70%的500bp插入物大小,20%的配偶对插入物和10%40000bp插入物。40000bp偏斜(skew)含有70%的40000bp插入物和10%500bp插入物。n50定义为50%的含有n50跨度的单体型区块的跨度。模拟重复10次并在y轴中记录平均n50。结果表明,单独的较高覆盖不能形成较长的单体型。此外,这些数据证明了较长的插入物大小片段生成较长的单体型。
实施例2:haploseq的实验策略
在haploseq中,发明人首先基于先前建立的hi-c实验方案进行了临近-连接测序(lieberman-aiden等,science326,289-293(2009))。与其他单体型分析方法中纯化的基因组dna相反,在从细胞分离dna之前,首先原位进行临近-连接(图1a)。特别地,空间临近的基因组区域原位交联,使用限制酶消化,再连接以形成人工片段,其接着被分离(图1a)。如此分离的纯化的dna片段可以捕获两个不同的基因组基因座,其在体内3d空间中圈在一起(dekker等,science295,1306-1311(2002);lieberman-aiden等,science326,289-293(2009);和kalhor等,naturebiotechnology30,90-98(2012))。确实,在对所得的dna文库进行鸟枪dna测序后,配对末端读出具有从几百个碱基对到数千万碱基对的范围的“插入物大小”,而其它方法倾向于生成范围从几百个到数万个碱基对的“插入物”(图1a-b)。理论上,haploseq中的实验方法保留了单体型信息,因为其允许同一条染色体上线性相距远的两个区域连接为短且连续的dna片段(图1a)。虽然hi-c实验中生成的短片段可以形成小的单体型区块,但长片段最终可以将这些小区块连接到一起(图1c)。使用足够的测序覆盖,这种方法允许连接不连续区块中的变体,并将每个这样的区块组装为单个单体型。因此,使用基于临近-连接的方法来制备dna测序文库,可以重构染色体跨度的单体型区块。
待考虑的一个因素是临近-连接可以捕获个体等位基因中的顺式相互作用和同源与非同源染色体之间的反式相互作用两者。虽然不同染色体之间的非同源的反式相互作用不影响定相,但同源染色体之间的反式相互作用(此后称为h-反式)可以使单体型重构复杂化,如果h-反式相互作用与顺式相互作用一样频繁。因此,发明人着手确定临近-连接测序数据中,h-反式相对顺式相互作用的相对频率。为了完成这一目的,发明人使用杂种小鼠胚胎干细胞(es)系,其衍生于两个近交纯合品系(小家鼠(cast)和129s4/svjae(j129))之间的杂交,对于所述细胞,亲本近交系全基因组序列(wgs)公开可用。作为结果,该细胞系中母本和父本单体型的知识作为育种结构的产物是先验已知的,且然后,等位基因之间的相互作用频率可以明确地检测。从这些杂种es细胞,发明人进行了hi-c实验并生成了超过6.20亿个可用的75碱基对配对末端读出,对应基因组的30x覆盖。
为了确定单体型内(顺式)相对于单体型间(h-反式)相互作用的程度,发明人使用先前的单体型信息来区分来自cast和j129等位基因的读出。为了检查h-反式相互作用模式,发明人首先可视化地检查了每个等位基因之间的相互作用模式(图2a)。先前的hi-c研究已经确认了长久以来建立的染色体领域的概念,尽管没有对每个染色体的两个等位基因之间进行区分(lieberman-aiden等,science326,289-293(2009);和kalhor等,naturebiotechnology30,90-98(2012))。发明人观察到每条染色体的cast和j129等位基因形成单独的染色体领域(图2a)。此外,在与顺式相互作用相比时,发明人观察到<2%的h-反式相互作用,表明绝大多数的hi-c读出真正是顺式(图2b)。另外,相对h-反式而言dna读出是顺式的概率似乎作为读出对之间插入物大小的函数而变化(图2c,和图6)。如图6所示,每个点描绘了lowess平滑曲线,而黑色的点源自于组合所有的染色体。这表明每条染色体遵循相似的h-反式相互作用概率的模式。这些观察表明,h-反式相互作用是罕见现象。
实施例3在杂种小鼠es细胞中以高分辨率精确重构染色体跨度的单体型
存在罕见的h-反式相互作用读出和诸如在变体位点处的测序误差等现象可导致同源配对之间错误的连接并引起单体型重构的冲突。为了克服这些问题,发明人将hapcut25软件整合到haploseq分析中以在概率上预测单体型。具体的是,hapcut构建了具有杂合变体作为节点和由重叠片段所阐明的边缘的图表。由于测序误差和h-反式相互作用,该图表可能含有几个假的边缘。hapcut使用最大切割算法来预测简约的(parsimonious)解决方案,其最大地与输入测序读出组提供的单体型信息一致(图3a)。因为临近-连接比常规基因组测序或配偶对产生更大的图表,发明人修改了hapcut以减少其计算时间,使其可用于haploseq分析。为了测试hapcut从临近-连接和测序数据生成单体型区块的能力,发明人再次利用了castx129小鼠es细胞hi-c数据。在这种情况下,发明人没有先验区分测序读出属于哪一个等位基因。取而代之,发明人允许hapcut从头重构杂合变体的单体型区块。接着,本发明人利用cast和j129等位基因的已知单体型信息来评估算法的性能。发明人使用了完全性,分辨率,以及精确度的度量来评估haploseq分析在单体型重构中的成功(图7)。
在图7a中,将杂合snp考虑为节点,且在属于相同片段的节点之间产生边缘。该图系统从头建立了两条同源染色体(或单体型)。然而,可以有形成的多个区块,且在本实施例中发明人已经鉴定了一个跨越96.15%的大型mvp组分以及不能被连接到mvp区块的另一个小型区块(示于黑色边缘的方框中)。
通过就跨越的碱基对的数目而言生成的单体型区块的大小或者通过每个区块跨越的杂合变体的总数测量单体型定相的完全性。一般来说,根据杂合变体连接,对于每条染色体,,hapcut将生成各种大小的几个单体型区块。含有定相的最杂合变体(mostheterozygousvariantphased)(mvp)的单体型区块通常是最感兴趣的,因为它经常是跨度最大的模块。另外,可以将少数杂合变体分配到较小的区块,由于它们不能与mvp区块连接。在这种情况下,mvp区块跨越了每条染色体的超过99.9%的可定相碱基对(图3b),证明使用hi-c数据的haploseq分析可以生成完全的染色体跨度的单体型。
虽然完全性定义为mvp区块的碱基对跨度,但分辨率被表示为定相的杂合变体相对于mvp区块中跨越的总变体的分数(图7)。对于每条染色体,生成的这些mvp区块具有高分辨率,因为发明人可以定相任意给定染色体上约95%的杂合变体(图3b)。不能将剩下的5%的杂合变体连接似乎是因为不存在覆盖这些变体的测序片段,或不能将这些杂合变体连接到mvp变体模块。作为结果,尽管跨越了绝大部分的染色体,mvp区块在定相的变体中含有约5%的缺口。
为了评估mvp区块中杂合变体的精确度,发明人将通过haploseq分析从头生成的预测的单体型与cast和j129等位基因的已知单体型比较。发明人将精确度定义为mvp区块中正确定相的定相杂合变体的分数(图7)。在分配到mvp单体型模块的变体中,在区分两个已知的单体型中发明人观察到了>99.5%的精确度(图3b)。
最后,由于发明人先前已经证明了h-反式相互作用概率随着分开两个测序读出的基因组距离而增加(图2c),本发明人将h-反式相互作用概率整合到hapcut算法中,并将测序读出的最大插入物大小加帽于3000万碱基对。这些条件不牺牲发明人生成的单体型的完全性。相反,发明人观察到mvp模块中变体进一步改进的精确性,伴随着定相的变体分辨率的中度下降(图8a和b)。
如这些图所示,限制性hapcut模型仅允许直到特定最大插入物大小(maxis)的片段。最低的maxis为500万碱基,在这之下mvp区组中形成染色体跨度的单体型的能力丧失。在较高的maxis,mvp区块的分辨率(a)高但含有较高的精确度(b)。因此,选择了3000万碱基作为maxis以允许可接受水平的分辨率和精确度。该模拟在低变体密度的情况下,在castxj129系统中的不同染色体中进行,因为这更接近人类应用。该分析没有整合h-反式概率,使得实现了maxis单独的效果。
总之,这些结果证明了对于所有的常染色体,haploseq分析产生完全的,高分辨率和精确的单体型。
实施例4haploseq与其他单体型定相方法比较
为了将本文公开的方法与先前建立的单体型分析方法比较,发明人模拟了常规配对末端鸟枪法dna测序(wgs),配偶对测序,fosmid和临近-连接的20x覆盖dna测序数据来评估每种方法重构单体型的能力。发明人观察到仅使用临近-连接的haploseq分析可以生成染色体跨度的mvp区块,而其它方法产生显著较小的mvp区块并因此具有片段化的单体型结构(图3c)。具体地,基于配偶对和fosmid的测序方法分别生成几十万碱基和约百万碱基大小的区块。发明人将wgs数据和配对,fosmid以及邻近连接组合,以增加覆盖并增加数据结构中的变异性,然而生成较长单体型的能力没有显著变化(图3c)。为了比较这些方法的分辨率,发明人检查了排名前100的经变体定相的单体型区块的累积调整跨度(图3d),其中调整的跨度表示为完全性和分辨率的乘积。haploseq中获得的单独mvp区块是完全的,并具有约90%的分辨率。相反,当将所有区块累积性考虑时,常规鸟枪法测序,配对和fosmid仅能覆盖5%,65%,和90%的染色体。与mvp区块的大小相比,累积完全性具有较少的潜在应用,因为不同区块中的变体彼此依然是未定相的。较高的覆盖(图3d,虚线)没有显著改变累积跨度模式。这表明为了生成染色体跨度的单体型模块,总测序覆盖似乎相比用于定相的方法不那样重要。
实施例5haploseq的表现依赖于变体密度
castxj129es细胞系的一个区别特征是整个基因组中存在高密度的杂合变体。平均每150个碱基存在一个杂合变体,其比人类中频繁7-10倍(wheeler等,nature452,872-876(2008)和pushkarev等,naturebiotechnology27,847-850(2009))。为了起始测试haploseq在人细胞中生成单体型的可行性,发明人对castxj129系统中的杂合变体进行了二次采样,使得变体密度模拟人群中的变体密度。发明人接着测试了多么低的变体密度影响haploseq重构单体型的能力。虽然降低的变体密度快速的降低了片段含有杂合变体的能力,但通过haploseq获得精确且完全的单体型区块的能力没有改变(图4b)。发明人仍然观察到了每个染色体里的完全单体型,而平均精确度仅少量降低,在低变体密度的情况下从约99.6%降到约99.2%(图4b)。然而,较低的变体密度的确导致了较少的可用读出,其继而提供较少的机会用于预测模型来解决单体型。作为结果,与高密度条件相比,使用“人类”变体密度生成的mvp区块具有较低的分辨率和较少的定相变体。现在mvp区块中约32%的杂合变体被定相(图4b),而不是高密度情况下的95%(图3b)。总的来说,低变体密度不影响完全性或精确度,但确实影响haploseq分析的染色体跨度的单体型的分辨率。
实施例6对人类个体的haploseq分析
为了实际地评估本文的方法在人类中定相单体型的能力,发明人在gm12878类淋巴母细胞细胞系中进行了haploseq。该细胞系的完全单体型已经由1000基因组计划从家庭三人组wgs确定15。发明人生成了超过2.62亿个可用的100碱基对配对末端读出,其对应约17x覆盖。在gm12878细胞中,haploseq成功地在所有的近端着丝粒染色体和18个中着丝粒染色体中的17个中生成了染色体跨度的单体型(图4c-d)。值得注意的是,先前尝试在人类中单体型重构的方法不能重构跨越中着丝粒染色体的高度重复的着丝粒区域的单体型(levy等,plosbiology5,e254(2007);kitzman等,naturebiotechnology29,59-63(2011);suk等,genomeresearch21,1672-1685(2011);duitama等,nucleicacidsresearch40,2041-2053(2012);和kaper等,procnatlacadsciusa110,5552-5557(2013))。使用haploseq,发明人生成了跨越除9号染色体外所有中着丝粒染色体的着丝粒的单体型,在9号染色体中错误的连接导致了着丝粒处单体型调用的切换。除了具有大型的15mbp不充分定位的着丝粒区域外,9号染色体具有相对较低的可用覆盖(13.7x)。发明人假设额外的覆盖可以提供跨越着丝粒的较好机会。因此,在9号染色体中,发明人将hi-c数据与先前生成的hi-c和tcc数据组合,这将其覆盖增加到约15x。栓系染色体捕获(tcc)与hi-c相似,其中交联的dna片段在固体表面栓系并连接到一起。tcc生成与hi-c实验相似的数据,具有稍微好一点的能力来捕获真正的长距离染色质相互作用(kalhor等,naturebiotechnology30,90-98(2012))。使用该组合的数据组,发明人能够精确地定相整个9号染色体。总的来说,从haploseq分析的仅17x基因组覆盖,发明人生成了所有人染色体(包括x染色体)的完全的,染色体跨度的单体型,尽管以降低的约22%分辨率(图4c)。
实施例7通过组合haploseq和局部条件性定相完全且高分辨率的单体型定相
虽然haploseq生成完全的染色体跨度的单体型,由于人类群体中的低变体密度,其不能达到高分辨率的定相的变体。这导致了“缺口”,其中杂合变体相对于mvp单体型区块仍然未定相。发明人推理使用衍生于群体规模测序数据的连锁不平衡模式(linkagedisequilibriumpatterns),可以将这些缺口变体以概率连接到mvp模块。为了这一目的,发明人使用beagle(v4.0)(browning等,genetics194,459-471(2013))软件和来自1000基因组计划的测序数据(genomesproject,c.等,nature491,56-65(2012)。发明人使用了haploseq生成的染色体跨度的单体型作为“种子单体型”以指导局部定相。作为结果,发明人能够从相对于mvp区块仍然未定相的“缺口”变体的连锁不平衡(ld)测量生成局部定相预测。
为了首先调查该方法的有效性,就mvp区块中定相的变体的数量而言,发明人以不同的分辨率百分比在gm12878基因组中模拟了染色体跨度的种子单体型。该模拟结果表明发明人可以甚至以低分辨率种子单体型输入时精确地推测局部定相(10%种子单体型分辨率时3%的误差,图5a中的上面曲线)。由于复杂的群体结构,来自beagle预测的局部单体型和haploseq种子单体型的定相预测之间发生偶尔的错配。为了修正这种现象,发明人检查了包围每个待推测杂合变体的临近窗口区域,并分析种子单体型和局部定相之间定相的一致。通过仅当它们具有100%的一致性时接受变体相对于所述种子单体型定相,不论单体型分辨率如何,发明人能够将误差率降低到约0.7%(图5a,下面的曲线)。由于该原因,发明人能够推测局部定相的杂合变体的分数随着较大的种子单体型分辨率而增加(图5a,底部图组)。发明人使用3个定相的种子单体型变体的邻域窗口大小,而窗口大小的增加不显著增加精确度。
基于这些结果,发明人使用来自haploseq分析生成的mvp染色体跨度的单体型作为种子单体型并进行了局部条件性定相。总体上,发明人以平均约98%的精确度生成了具有约81%分辨率的染色体跨度的单体型(图5b)。值得注意的是,在不能局部定相的19%的杂合变体中,约16%是由于它们不存在于群体样品中而约3%是因为邻域校正,其仅少量地影响分辨率(图5b)。因此,通过组合haploseq分析和局部条件性定相,发明人能够在人类中完成高分辨率和精确的染色体跨度的单体型。
实施例8通过haploseq获得精确和高分辨率的染色体跨度的单体型的要求
从局部条件性定相分析,发明人推导具有约20-30%分辨率的种子单体型足以获得精确和高分辨率的染色体跨度的单体型。因此,接下来的问题是完成具有约20-30%分辨率的染色体跨度的种子单体型的最小实验要求是什么。为了研究这点,发明人生成了模拟的临近-连接测序数据,其具有不同的读出长度和测序覆盖。基于模拟,为了首先完成染色体跨度的单体型,依赖于获取约15x的可用测序覆盖而不论读出长度如何(图5c)。在获得染色体跨度的单体型后,完成所需的约20-30%的分辨率的分数将要求约25-30x可用覆盖,其具有100个碱基对配对末端读出(图5d)。该模拟还强调对较长读出长度的需要,因为较长的读出长度显著增加种子单体型分辨率。另外,该模拟不考虑精确度,且从对gm12878的分析,发明人能够推导重构精确单体型的能力依赖于可用覆盖。例如,低覆盖染色体例如17号和19号具有相对较低的精确度。具体的是,较低的覆盖可能导致许多变体以较少的边缘连接,其继而可以传播(propagate)高度误差的结构到整个染色体跨度的单体型。见以下表1。
表1示出了mvp区块的覆盖和精确度之间的关系。低覆盖影响临近-连接完成精确单体型的能力,正如在17号,19号和20号染色体中看到的。局部条件性定相(lcp)后,分辨率从22%增加到81%(图5b)而不进一步降低精确度。实际上,基于邻域校正看到了精确度的少量增加。如图5b中还示出的,最后一栏反映总体精确度。
此外,虽然对于任意一个染色体,发明人没有达到约25x的可用覆盖,发明人仍然能够达到平均约~98%的精确度。额外的覆盖可以甚至进一步增加精确度,正如在低密度castxj129系统中观察到的。因此,具有100个碱基对的配对末端读出的25-30x可用覆盖足以完成具有约20-30%分辨率的染色体跨度的单体型,并允许使用haploseq分析精确的局部条件性定相。
表1
实施例9对人类个体的haploseq分析
在本实施例中,使用来自四个人类个体的样品进行haploseq分析。为了该目的,在甲醛交联之前,将人组织样品急速冷冻并研磨。接着,如lieberman-aiden等,science326,289-293(2009)所述在样品上进行hi-c。使用先前描述的haploseq方法进行单体型分析(selvaraj等,natbiotechnol.2013dec;31(12):1111-8)。简单的说,为了生成单体型预测,来自每个供体的hi-c读出用作对hapcut软件的输入测序(bansal等,bioinformatics.2008aug15;24(16):i153-9)。对于最终单体型调用,将hi-c数据与供体基因组的wgs配偶对数据组合。因为hi-c数据仅能定相一些snp,通过利用来自1000基因组工程的群体测序数据进行局部条件性定相方法。对于每条染色体,haploseq生成两个单体型,对于母本等位基因为1个,对于父本等位基因为1个。一个等位基因命名为p1(亲本1),而另一个等位基因命名为p2(亲本2),因为关于每个供体基因组中的来源亲本的信息不可用。
对于四个不同的组织供体,在每个组织供体中发明人能够生成跨越整个染色体的单体型,其具有平均99.5%的完全性(单体型解决的基因组区域的覆盖),以及范围从78%至89%的平均分辨率(定相的杂合snp的覆盖)。通过比较预测的单体和属于相同配对末端读出的snp的一致(concordance),验证单体型预测的精确度。对于h3k27acchip-seq读出,一致率为99.7%而对于mrna-seq读出为98.4%,表明高度的精确度。
实施例10使用捕获-hic和测序的靶向单体型分析
在本实施例中,具有寡核苷酸探针的捕获-hic用于捕获染色质相互作用,用于对整个人hla基因座的靶向单体型分析。
为了生成hi-c文库,将gm12878(coriell)细胞在使用15%fbs和1x青霉素/链霉素补充的85%的rpmi培养基中悬浮培养。收获gm12878细胞,甲醛固定,并经过如lieberman-aiden等,science326,289-293,(2009)中所述hi-c实验方案,在捕获测序前使用一些修改。在将illumina衔接头(adapter)连接到hi-c片段上后,文库经历14个pcr扩增循环,接着使用高保真(fusion)聚合酶捕获杂交。可以根据需要多少dna用于下游捕获杂交反应,修改预捕获pcr循环的数量。在这种情况下,使用少量珠子结合的hi-c文库输出以14个循环进行几个平行的pcr反应以最大化pcr产出和获得足够的材料用于可再生捕获-hic实验。为了检查文库质量和为了提供内部深度匹配的对照用于捕获-hic文库,在预捕获(14个循环)文库上进行ngs。
使用上文所述的实验方案,首先生成了具有足够材料的常规hi-c文库,以使得能够进行基于寡核苷酸探针的整个hla区域的捕获(图9和图10a)。
为了获得人hla基因座的靶向单体型分析,计算生成寡核苷酸探针序列并靶向hla基因座中靠近hindiii切割位点的非重复+/-400bp区域(图10)。为此,进行了单体型分析表现模拟。简单的说,根据单体型分析分辨率(y轴)作为测序覆盖(x轴)的函数模拟haploseq表现。进行了该研究来更普遍提问如果在文库中仅存在含有hindiii切割位点相邻序列的hi-c片段,haploseq将表现得多好。理论上来说,捕获-hic文库将仅含有其中至少一个读出末端起源于hindiii切割位点相邻序列的hi-c片段。因此,使用内部的常规hi-c数据组,使用所有定位的hi-c读出进行haploseq分析,而没有限制任何读出(resolution_nores)。可用读出也被限制为仅那些含有在hindiii切割位点的500bp以内(resolution_pm500)或切割位点的250bp以内(resolution_pm250)的至少1个读出末端。该模拟的结果表明,虽然单体型分析分辨率有约20%的降低,该分辨率仍然足够用于单体型分析的目的。该结果还表明,无论将读出限制到靠近hindiii切割位点的250bp还是500bp,分辨率存在少量差异。相应地,选择400bp用于靶向方法。
使用suredesign参数,以4x铺盖密度在靶向区域处设计探针,以优化捕获效率和因此最大化单体型分析分辨率和精确度。更具体的是,为了生成rna诱饵,使用suredesign软件包(agilenttechnologies)设计探针。使用hg19基因组构建(chr6:29689001-33098938),定制设计靶向靠近hindiii切割位点的上游和下游400bp,跨越mhc基因座。将suredesign参数设置为4x铺盖密度,最大探针加强(boosting),以及最大重复序列掩盖。尽管不临近hindiii切割位点,发明人也以2x铺盖密度,平衡加强,以及最大重复元件掩盖靶向hla基因外显子。通过suredesign使用本文所述的参数,总共计算生成了12,298个探针。
接着,通过customarrayinc合成了单链dna(ssdna)寡聚物。ssdna寡聚物含有通用的正向和反向引发序列。正向引发序列包含截短的sp6rna聚合酶识别序列。反向通用引发序列含有bsrdi识别序列,用于体外转录前3’剪切。为了将寡聚物转化为生物素化的rna诱饵,稀释寡聚物并接着使用高保真dna聚合酶(kapa)进行pcr扩增,并接着柱纯化(promega)。该pcr反应也用于填充到sp6识别序列的剩余部分。接着,通过使用bsrdi(newenglandbiosciences)消化dsdna去除反向引发序列并再次纯化以去除消化的片段。最后,根据制造商的方案(ambion)在生物素化的utp(epicentre)存在下进行体外转录(ivt)。接着,柱纯化rna(qiagen),稀释到工作浓度(500ng/μl)并在使用前储存在-80℃。
为了富集hi-c文库用于将hi-c片段定位到hla基因座,主要根据customarray实验方案和一些修改进行捕获杂交以及接着pcr扩增。简单的说,将500nghi-c文库在65℃与500ng生物素化的rna探针孵育过夜。因为靶向序列(约320kb)仅为基因组的约0.01%,发明人每个实验进行了16个平行的杂交反应,并在测序前将最终杂交产物汇集。接着,使用链霉亲合素包被的珠子(invitrogen)下拉rna:dna杂交体,未结合的dna片段被洗掉,并洗脱捕获的产物。在捕获的产物被洗脱后,将它们在qiagenminelute柱上脱盐,并使用11个循环pcr扩增(fusion)。在该实验方案中,对于每个杂交反应,所有的步骤独立地进行。换句话说,在脱盐的捕获片段上进行几个平行的捕获后pcr反应,且每种捕获后pcr产物使用ampurexp珠子(beckmancoulter)独立地纯化。接着,汇集pcr产物并接着使用真空离心蒸发浓缩器(speed-vac)浓缩。得到的捕获-hic文库,接着在illuminahiseq2500上进行下一代测序。
更具体的是,在制备捕获hi-c文库后,所得的文库以约1x测序深度测序,使用配对末端100bp读出长度。理论上来说,该测序深度将足够覆盖基因组中的每个碱基一次。接着计算整个hla基因座中的覆盖(包括跨越基因座的所有非靶向序列)并确定为约32.1x。为了计算hla基因座富集,将hla覆盖除以基因组覆盖。将所有来自捕获-hic测序数据的单克隆定位读出二进制化到100kb二进制全基因组。这里,将落在hla基因座以及6号染色体上靠近的脱靶区域的每个二进制中的读出总数作图。发现靶向hla基因座为约29m至33.4m,其相对于6号染色体上的非靶向靠近区域表现出显著的富集。
总的来说,通过在hi-c文库上进行上述的捕获测序,生成了gm12878人类类淋巴母细胞细胞系(lcl)的捕获-hic文库,在hla基因座上以约1.1x测序深度以约30倍的富集生成。
由于单体型分析效率取决于3d染色体接触的保真度,调查了与常规hi-c文库相比,在相同的基因座捕获-hic数据组是否保留了相对接触频率。为了该目的,将来自捕获hi-c的染色质相互作用与先前发表的来自gm12878细胞的hla基因座的hi-c数据比较。简单的说,使用来自gm12878的捕获-hic数据(顶部),以及来自gm12878的发表数据(selvaraj等,natbiotechnol.2013dec;31(12):1111),以20kb二进制生成了hla基因座中的接触矩阵。在生成接触矩阵前,每个数据组通过读出深度标准化,其简单地以每个矩阵值(i,j)除以定位到基因座的读出的总数。发现在这些数据组中存在高度显著的一致性(p<0.01)。
除了检查捕获-hic数据中是否保留了相对3d接触频率外,也进行了测定以更接近的检查hi-c片段的特性。首先,使用所有的捕获-hic数据(包括通过所述实验捕获的脱靶序列),发明人比较了捕获和常规hi-c文库中的染色体内(顺式)和染色体间(反式)读出的比例,并发现顺式:反式的比例彼此一致。第二,如果每个数据组被限制到仅定位到hla基因座的读出,再次发现每个数据组含有大体相同的顺式:反式比例。第三,由于haploseq关键地取决于同一条同源染色体中高频率的顺式接触(h-顺式)(~99%),探索了捕获-hic数据中的h-顺式率。发现捕获-hic数据也含有绝大多数(约98%)的h-顺式hi-c片段,因此能够进行有效的haploseq分析。该分析揭示了常规hi-c和捕获-hic文库一般具有可比的顺式:反式比率且捕获-hic具有相似的同源性-反式相互作用,因此保留了单体型内接触频率,其对于使用haploseq保持高单体型分析精确度至关重要。
另外,进行了对捕获-hicrna探针灵敏度的分析。作为评价捕获-hic探针表现的指标,发明人分析了每个探针序列中的读出密度以及具有至少1个捕获的hi-c片段的探针总分数。为了该目的,将读出密度(y轴)相对于每个独特的rna探针序列(x轴)作图以生成柱状图。该柱状图中每个垂直线代表单个独特的探针。发现在总共7885个独特探针中,7650个(~97%)具有至少一个读出定位到由该探针靶向的序列。这提供了捕获测序方法的总体灵敏度的某些意义。
总的来说,以上结果示出了捕获-hic实验方案数据是高质量数据,因此能够对单体型模型进行精确的分析。
接着,使用haploseq(selvaraj等,natbiotechnol.2013dec;31(12):1111-8)和lcp实验方案从捕获-hic数据进行了单体型重构。首先,从先前公开的数据获得了gm12878的定相信息(genomesproject,c.等,nature467,1061-1073,(2010))。接着,利用haploseq和局部条件性定相(lcp)实验方案来生成hla基因座里的单个单体型结构,并定相gm12878中~95%的等位基因。将haploseq分析的单体型结果总结于以下表格中。接着,将预测的单体型结构与先前报道的单体型结构比较并估算捕获-hic的精确度为~97.7%(见以下表2)。
表2
如该表格中所示,在hapcut后,发明人生成了hla基因座的完全单体型结构,并以约96%精确度定相了约46%的所有杂合snp。lcp后,以约98%精确度定相了约95%的所有的杂合snp。在最终的单体型结构中,发现由hapcut和lcp定相的snp的精确度分别为约96%和99%。
值得注意的是,本文公开的方法是首次证明了跨越整个hla基因座的高质量单体型分析,在单一单体型结构中不仅定相高度不同的主要和次要hla等位基因基因座,而且还定相一起跨越基因座的其它重要的免疫学基因和非hla基因座。更广泛的说,这种方法学是首次实现了使用者确定的靶向基因座的完全单体型结构(kaper等,procnatlacadsciusa110,5552-57(2013))。通过对于95%的等位基因完成精确的单体型(约98%),这一方法可以用于个体化基因组学和群体遗传学。
以上实施例和优选实施方案的说明应当认为是说明性的,而不限制如权利要求所确定的本发明。如将容易理解的是,可以利用上述特征的众多变化和组合而不脱离如权利要求中所阐明的本发明。这些变化不视为脱离本发明的范围,且所有这些变化意图包括在以下权利要求的范围内。本文引用的所有参考文献以其整体并入本文。
- 还没有人留言评论。精彩留言会获得点赞!