一种利用过渡金属催化咔唑与芳基肼反应合成N-芳基咔唑类化合物的方法与流程
本发明涉及有机合成技术领域,具体涉及一种利用过渡金属催化咔唑与芳基肼反应合成n-芳基咔唑类化合物的方法。
背景技术:
咔唑类化合物是一类重要的有机含氮杂环化合物,普遍存在于自然界中,在医药、染料和有机光电材料等领域中有着广泛地应用,在人类日常生活中具有重要的意义。咔唑类化合物具有抗癌、抗菌、抗精神病、抗分裂、抗氧化等多种药理活性,例如,咔唑霉素b7具有非常优异的抗炎特性,椭圆胺衍生物可以很好地抑制癌细胞的生长。此外,这些杂芳香族化合物在材料科学中也有许多应用,由于含咔唑的聚合物具有良好的空穴迁移率和光电导率,其在有机发光材料的发展中起着不可替代的作用,含有咔唑的聚合物已在复印机、激光打印机和全息防伪印章等许多设备中商业化。咔唑类材料由于具有形成稳定的自由基阳离子(空穴)的能力,通常表现出良好的电荷载流子能动性,常被用作空穴输运材料。
2011年,niranjanpanda课题组报道了一种高效的铁酸铜催化的含氮杂环和芳香卤代物形成碳氮键的反应,以dmf为溶剂[n.panda/tetrahedronletters,2011.52,1924-1927],但是高沸点溶剂的使用使得后处理较难。2018年,luigivaccaro课题组报道了一种糠醇与水的共沸物为溶剂,采用碘化亚铜催化的含氮杂环与芳香卤代物偶联的方法来制备n-芳基化合物[l.vaccaro/greenchem,2018.20,1634-1639],该方法绿色环保,为n-芳基化合物的制备提供了一种高效的方法,但是该反应的反应温度高、时间长,反应所生成的含卤副产物污染环境。
在传统的有机合成中,n-芳基咔唑类化合物是通过铜催化的乌尔曼偶联反应制备的,即芳基卤代物与咔唑环的直接偶联。这类反应往往需要较高的反应温度,由于很多官能团对温度的耐受性较差,因而该类反应的官能团容忍性相对较差,而且该反应需要当量的铜作催化剂、反应时间较长,这在一定程度上造成了环境负担,提高了合成成本。
技术实现要素:
本发明提供了一种利用过渡金属催化咔唑与芳基肼反应合成n-芳基咔唑类化合物的方法,以解决目前咔唑类化合物合成过程中后处理难、污染环境或者官能团容忍性差的问题。
本发明所采用的技术方案是:一种利用过渡金属催化咔唑与芳基肼反应合成n-芳基咔唑类化合物的方法,以过渡金属盐为催化剂,以咔唑类化合物和芳基肼类化合物为反应底物,将反应底物、碱类化合物添加到溶剂中经搅拌加热,反应一定时间后得到反应液,对反应液分离提纯得到n-芳基咔唑类化合物。
优选地,过渡金属盐的金属中心为pd、cu、fe、ni、mn、co中的任意一种。
优选地,芳基肼类化合物选自苯肼、2-溴苯肼、4-溴苯肼、4-甲基苯肼、联苯-3-肼、联苯-4-肼、3-溴苯肼中的任意一种。
优选地,咔唑类化合物选自咔唑、1-苯基咔唑、1-溴咔唑、2-溴咔唑、3-溴咔唑、4-溴咔唑中的任意一种。
优选地,所述的溶剂为乙腈,该反应在空气的气体氛围下进行。
优选地,反应温度为0-60℃、反应时间为4-12h。
优选地,碱类化合物选自碳酸铯、碳酸钾、氢氧化钠、氢氧化钾、氟化钠、氟化钾、氟化铯、dbu和tmeda中的任意一种。
优选地,咔唑类化合物与碱类化合物用量的摩尔比为:1:(1-10)。
优选地,咔唑类化合物与芳基肼类化合物用量的摩尔量为1:(1-5),咔唑类化合物与过渡金属盐用量的摩尔量比为1:(0.1-10)。
优选地,向反应液中加入饱和食盐水,用二氯甲烷萃取三次,并加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将溶剂除去并经过柱得到n-芳基咔唑类化合物。
本发明与现有技术相比具有以下优点:本发明采用低沸点乙腈作为反应溶剂,反应后处理易于进行;以空气氧作为氧化剂,反应条件绿色环保、安全、成本低,避免过度氧化;以芳基肼类化合物作为芳基化试剂,反应副产物只有水和氮气,环境友好无污染;以过渡金属盐为催化剂,其活性高,避免了当量贵金属的使用,降低了合成成本,避免了传统合成方法所带来的弊端;此外,该方法的反应温度低,因而其对官能团的容忍性相对较好。总的来说,该合成方法绿色环保、工艺简捷、反应选择性好、产率高,具有较强的工业应用前景。
附图说明
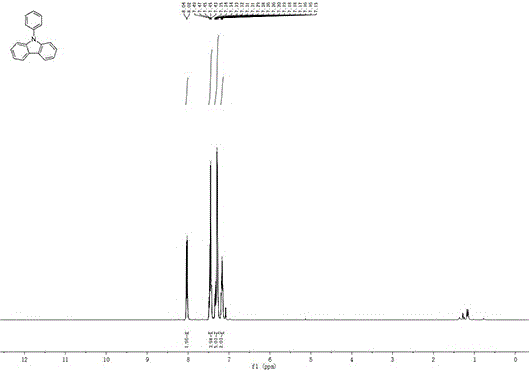
图1是实施例1制备的n-苯基咔唑的1hnmr谱图;
图2是实施例1制备的n-苯基咔唑的13cnmr谱图;
图3是实施例2制备的4-溴-n-苯基咔唑的1hnmr谱图;
图4是实施例2制备的4-溴-n-苯基咔唑的13cnmr谱图;
图5是实施例3制备的9-[1,1'-联苯-4-基]-3-溴-9h-咔唑的1hnmr谱图;
图6是实施例3制备的9-[1,1'-联苯-4-基]-3-溴-9h-咔唑的13cnmr谱图;
图7是实施例4制备的9-[1,1'-联苯]-3-基-2-溴-9h-咔唑的1hnmr谱图;
图8是实施例4制备的9-[1,1'-联苯]-3-基-2-溴-9h-咔唑的13cnmr谱图;
图9是实施例5制备的9-(2-溴苯基)咔唑的1hnmr谱图;
图10是实施例5制备的9-(2-溴苯基)咔唑的13cnmr谱图;
图11是实施例6制备的9-[1,1'-联苯]-3-基-3-溴-9h-咔唑的1hnmr谱图;
图12是实施例6制备的9-[1,1'-联苯]-3-基-3-溴-9h-咔唑的13cnmr谱图;
图13是实施例7制备的9-(4-溴苯基)咔唑的1hnmr谱图;
图14是实施例7制备的9-(4-溴苯基)咔唑的13cnmr谱图;
图15是实施例8制备的2-溴-9-苯基-9h-咔唑的1hnmr谱图;
图16是实施例8制备的2-溴-9-苯基-9h-咔唑的13cnmr谱图;
图17是实施例9制备的3-溴-n-苯基咔唑的1hnmr谱图;
图18是实施例9制备的3-溴-n-苯基咔唑的13cnmr谱图;
图19是实施例10制备的n-芳基咔唑类化合物的1hnmr谱图;
图20是实施例10制备的n-芳基咔唑类化合物的13cnmr谱图;
图21是实施例11制备的3-溴代-n-甲苯基咔唑的1hnmr谱图;
图22是实施例11制备的3-溴代-n-甲苯基咔唑的13cnmr谱图;
图23是实施例12制备的9-(3-溴苯基)-9h-咔唑的1hnmr谱图;
图24是实施例12制备的9-(3-溴苯基)-9h-咔唑的13cnmr谱图;
图25是实施例13制备的1-溴-n-苯基咔唑的1hnmr谱图;
图26是实施例13制备的1-溴-n-苯基咔唑的13cnmr谱图。
具体实施方式
为了对本发明进行更好地说明,现结合实例对其进行进一步的说明。
一种利用过渡金属催化咔唑与芳基肼反应合成n-芳基咔唑类化合物的方法,其特征在于:以过渡金属盐为催化剂,以咔唑类化合物和芳基肼类化合物为反应底物,将反应底物和碱类化合物添加到溶剂中,经搅拌加热后,反应一定时间得到反应液,对反应液经萃取、柱层析分离提纯得到n-芳基咔唑类化合物,其中过渡金属盐的金属中心为pd、cu、fe、ni、mn、co中的任意一种,芳基肼类化合物选自苯肼、2-溴苯肼、4-溴苯肼、4-甲基苯肼、联苯-3-肼、联苯-4-肼中的一种,咔唑类化合物选自咔唑、1-苯基咔唑、2-溴咔唑、3-溴咔唑、4-溴咔唑中的一种,所述的溶剂为乙腈,该反应在空气的气体氛围下进行,以空气氧作为氧化剂,安全环保、成本低廉,反应温度为0-60℃、反应时间为4-12h。碱类化合物选自碳酸铯、碳酸钾、氢氧化钠、氢氧化钾、氟化钠、氟化钾、氟化铯、dbu和tmeda中的任意一种,咔唑类化合物、芳基肼类化合物、碱类化合物、过渡金属盐用量的摩尔比为:1:(1-5):(1-10):(0.1-10)。
实施例1
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物苯肼0.2mmol(22mg),咔唑0.2mmol(33mg),氯化铜0.05mg(0.1mol%),碳酸铯130mg(4eq.),乙腈2ml,加入磁子,0℃搅拌反应4h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为99%。目标产物表征数据:colorlessoil.(hexane:dcm=4:1aseluent).1hnmr(400mhz,cdcl3)δ=8.23(d,j=7.8hz,2h),7.71–7.60(m,4h),7.56–7.51(m,1h),7.51–7.46(m,4h),7.41–7.33(m,2h).13cnmr(100mhz,cdcl3)δ=140.97,137.79,129.94,127.51,127.22,126.00,123.43,120.38,119.98,109.84.
实施例2
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物苯肼0.6mmol(65mg),4-溴咔唑0.2mmol(50mg),碘化亚铜2mg(5mol%),碳酸铯130mg(4eq.),乙腈2ml,加入磁子,0℃搅拌反应4h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为95%。目标产物表征数据:whitesolid.(hexane:dcm=2:1aseluent).1hnmr(600mhz,cdcl3)δ8.93(d,j=7.9hz,1h),7.66(t,j=7.7hz,2h),7.61–7.49(m,5h),7.42(t,j=8.0hz,2h),7.37(d,j=8.1hz,1h),7.28(t,j=7.9hz,1h).13cnmr(150mhz,cdcl3)δ142.24,141.35,137.19,130.06,128.09,127.59,126.67,126.38,124.11,122.90,122.79,122.04,120.06,116.77,109.65,108.74.
实施例3
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物联苯-4-肼0.2mmol(44mg),3-溴咔唑0.2mmol(50mg),氯化亚铜1mg(5mol%),碳酸钾55mg(2eq.),乙腈2ml,加入磁子,0℃搅拌反应4h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为95%。目标产物表征数据:whitesolid.(hexane:dcm=3:1aseluent).1hnmr(600mhz,cdcl3)δ8.33(d,j=1.7hz,1h),8.18–8.14(m,1h),7.85(d,j=8.3hz,2h),7.74(d,j=7.8hz,2h),7.62(d,j=8.3hz,2h),7.59–7.54(m,3h),7.54–7.46(m,3h),7.42–7.35(m,2h).13cnmr(150mhz,cdcl3)δ141.26,140.68,140.17,139.58,136.42,129.08,128.72,128.65,127.84,127.28,127.20,126.80,125.26,123.15,122.44,120.60,120.49,112.86,111.41,110.16.
实施例4
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物联苯-4-肼0.2mmol(44mg),2-溴咔唑0.2mmol(50mg),硝酸铜3mg(7mol%),碳酸钾55mg(2eq.),乙腈2ml,加入磁子,0℃搅拌反应4h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为92%。目标产物表征数据:whitesolid.(hexane:dcm=3:1aseluent).1hnmr(600mhz,cdcl3)δ8.17(d,j=7.7hz,1h),8.04(d,j=8.2hz,1h),7.86(d,j=8.0hz,2h),7.74(d,j=7.4hz,2h),7.68–7.62(m,3h),7.56(t,j=7.2hz,2h),7.48(dd,j=17.8,5.7hz,4h),7.41–7.35(m,1h).13cnmr(150mhz,cdcl3)δ141.75,141.15,140.87,140.18,136.23,129.04,128.74,127.82,127.37,127.22,126.48,123.22,122.89,122.43,121.56,120.54,120.38,119.66,112.95,110.11.
实施例5
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物2-溴苯肼0.6mmol(112mg),咔唑0.2mmol(34mg),醋酸锰4mg(7mol%),氟化钠84mg(10eq.),乙腈2ml,加入磁子,30℃搅拌反应4h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为92%。目标产物表征数据:whitesolid.(hexane:dcm=5:1aseluent).1hnmr(600mhz,cdcl3)δ8.26(s,2h),7.95(d,j=7.2hz,1h),7.63–7.31(m,7h),7.19(d,j=7.9hz,2h).13cnmr(150mhz,cdcl3)δ140.98,136.88,134.33,131.23,130.24,128.91,126.06,123.95,123.38,120.46,120.12,110.14.
实施例6
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物联苯-3-肼1mmol(184mg),3-溴-咔唑0.2mmol(50mg),碳酸锰4mg(2mol%),氟化钾116mg(10eq.),乙腈2ml,加入磁子,30℃搅拌反应8h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为91%。目标产物表征数据:whitesolid.(hexane:dcm=4:1aseluent).1hnmr(600mhz,cdcl3)δ8.34(d,j=1.9hz,1h),8.16(d,j=7.8hz,1h),7.82(d,j=1.6hz,1h),7.78–7.75(m,1h),7.71(dd,j=12.2,7.8hz,3h),7.58–7.49(m,6h),7.45(dd,j=7.7,6.3hz,1h),7.40–7.36(m,2h).13cnmr(150mhz,cdcl3)δ143.33,141.30,140.00,139.63,137.84,130.45,129.07,128.75,128.03,127.22,126.82,126.49,125.74,125.67,125.25,123.17,122.43,120.61,120.49,112.86,111.39,110.14.
实施例7
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物4-溴苯肼1mmol(187mg),咔唑0.2mmol(34mg),醋酸钯4mg(10mol%),氟化铯300mg(10eq.),乙腈2ml,加入磁子,60℃搅拌反应12h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为96%。目标产物表征数据:whitesolid.(hexane:dcm=2:1aseluent).1hnmr(600mhz,cdcl3)δ8.23(d,j=7.7hz,2h),7.79(d,j=7.8hz,2h),7.50(t,j=7.6hz,4h),7.46(d,j=8.1hz,2h),7.39(t,j=7.3hz,2h).13cnmr(150mhz,cdcl3)δ140.71,136.90,133.19,128.78,126.20,123.62,120.96,120.51,120.35,109.66.
实施例8
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物苯肼0.6mmol(65mg),2-溴咔唑0.2mmol(50mg),肽菁钴11mg(10mol%),tmeda93mg(4eq.),乙腈2ml,加入磁子,60℃搅拌反应12h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为98%。目标产物表征数据:whitesolid.(hexane:dcm=3:1aseluent).1hnmr(600mhz,cdcl3)δ8.17(s,1h),8.03(d,j=5.0hz,1h),7.67(s,2h),7.57(dd,j=28.5,8.5hz,4h),7.52–7.41(m,3h),7.36(s,1h).13cnmr(150mhz,cdcl3)δ141.78,141.19,137.15,130.13,127.97,127.18,126.44,123.16,122.83,122.37,121.54,120.48,120.36,119.62,112.88,110.04.
实施例9
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物苯肼0.6mmol(65mg),3-溴咔唑0.2mmol(50mg),氯化镍3mg(10mol%),dbu122mg(4eq.),乙腈2ml,加入磁子,60℃搅拌反应12h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为96%。目标产物表征数据:whitesolid.(hexane:dcm=3:1aseluent).1hnmr(600mhz,cdcl3)δ8.30(s,1h),8.13(d,j=7.8hz,1h),7.65(t,j=7.7hz,2h),7.56(d,j=7.4hz,2h),7.54–7.51(m,2h),7.48(t,j=7.6hz,1h),7.44(d,j=8.1hz,1h),7.35(t,j=7.3hz,1h),7.30(t,j=8.7hz,1h).13cnmr(150mhz,cdcl3)δ141.29,139.62,137.31,130.03,128.64,127.81,127.09,126.71,125.16,123.08,122.33,120.52,120.37,112.73,111.30,110.05.
实施例10
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物苯肼0.6mmol(65mg),1-苯基咔唑0.2mmol(49mg),氯化铁3mg(10mol%),koh110mg(4eq.),乙腈2ml,加入磁子,0℃搅拌反应4h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为94%。目标产物表征数据:whitesolid.(hexane:dcm=4:1aseluent).1hnmr(600mhz,cdcl3)δ8.22(d,j=7.4hz,2h),7.86(d,j=7.4hz,2h),7.75(d,j=7.1hz,2h),7.69(d,j=7.3hz,2h),7.55(dd,j=17.6,8.2hz,4h),7.51–7.44(m,3h),7.36(t,j=6.8hz,2h).13cnmr(150mhz,cdcl3)δ140.93,140.35,140.33,136.93,129.00,128.54,127.69,127.38,127.19,126.01,123.49,120.37,120.03,109.89.
实施例11
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物4-甲基苯肼1mmol(122mg),3-溴咔唑0.2mmol(49mg),硫酸亚铁3mg(10mol%),k2co3110mg(4eq.),乙腈2ml,加入磁子,60℃搅拌反应12h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为94%。目标产物表征数据:whitesolid.(hexane:dcm=2:1aseluent).1hnmr(600mhz,cdcl3)δ8.29(d,j=1.5hz,1h),8.13(d,j=7.8hz,1h),7.51(dd,j=8.7,1.5hz,1h),7.48–7.43(m,5h),7.41(d,j=8.2hz,1h),7.33(t,j=7.4hz,1h),7.28(d,j=8.6hz,1h),2.53(s,3h).13cnmr(150mhz,cdcl3)δ141.44,139.78,137.78,134.59,130.61,128.56,126.93,126.63,125.03,123.02,122.22,120.47,120.19,112.54,111.31,110.07,21.28.
实施例12
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物3-溴苯肼0.6mmol(129mg),咔唑0.2mmol(34mg),硫酸亚铁3mg(10mol%),koh110mg(4eq.),乙腈2ml,加入磁子,60℃搅拌反应12h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为94%。目标产物表征数据:whitesolid.(hexane:dcm=2:1aseluent).1hnmr(600mhz,cdcl3)δ8.18(d,j=7.8hz,2h),7.79(s,1h),7.64(d,j=7.9hz,1h),7.57(d,j=7.9hz,1h),7.51(t,j=7.9hz,1h),7.49–7.44(m,4h),7.35(t,j=7.0hz,2h).13cnmr(150mhz,cdcl3)δ140.60,139.17,131.16,130.56,130.19,126.17,125.78,123.58,123.26,120.43,120.38,109.66.
实施例13
结构式如下的n-芳基咔唑类化合物的制备方法:
向50ml史莱克管中加入底物苯肼0.6mmol(65mg),1-溴咔唑0.2mmol(50mg),硫酸亚铁3mg(10mol%),naoh110mg(1eq.),乙腈2ml,加入磁子,40℃搅拌反应12h。反应结束后,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。反应液旋蒸后过柱得到目标产物,收率为94%。目标产物表征数据:whitesolid.(hexane:dcm=2:1aseluent).1hnmr(600mhz,cdcl3)δ8.16(d,j=7.7hz,2h),7.62(d,j=7.7hz,1h),7.61–7.56(m,3h),7.49(d,j=7.3hz,2h),7.44(t,j=7.7hz,1h),7.35(t,j=7.4hz,1h),7.18(t,j=7.7hz,1h),7.13(d,j=8.2hz,1h).13cnmr(150mhz,cdcl3)δ143.28,137.95,137.70,131.06,130.15,129.00,128.63,126.65,126.32,122.43,120.80,120.51,120.12,119.45,110.59,103.
按照实施例1的方法合成n-苯基咔唑类化合物,其各种反应条件和反应结果见表1。
表1不同条件下合成的n-芳基咔唑类化合物的情况
由以上实施例可知,采用本发明的制备方法,收率均达到了90%以上,反应效率高。
以上所述的仅是本发明的优选实施例,应当指出,对于本领域的技术人员来说,在不脱离本发明整体构思前提下,还可以作出若干改变和改进,这些也应该视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!