靶向敲除GLK基因的CRISPR/Cas9系统及其应用
本发明涉及分子生物学技术领域,涉及一种靶向敲除glk基因的crispr/cas9系统及其应用。
背景技术:
glk转录因子促进许多核编码的光合作用基因的表达,这些基因与叶绿素的生物合成有关。glk基因已经参与了拟南芥和苔藓小叶藓(physcomitrellapatens)叶绿体发育的调控。此外,异位表达glks能够诱导拟南芥、水稻根和愈伤组织等非绿色组织中叶绿体的产量增加,提高番茄中细胞叶绿体数量,表明glks在质体发育中的重要作用。冮慧欣等在bpglk基因功能的进行了更深入的研究,主要鉴定了t-dna插入突变株的表型和突变基因,总结了t-dna整合模式,研究了突变基因bpglk的功能,叶片颜色形成的机制及其机理。研究结果可为glk基因在木本植物中的应用提供基础,使人类能够利用分子工程技术创造出黄叶特色性状树,以满足园林绿化的需要。同时,白桦bpglk抑制的表达谱系也可为提供黄色高大乔木资源用于美化环境。
以往在开展植物crispr/cas9定点编辑技术中,人们主要利用根癌农杆菌介导法在t-dna的驱动下,将cas9蛋白-grna核糖核蛋白复合物(rnp)导入受体细胞,结果在获得的基因组编辑植物宿主基因组中仍含有外源dna序列(根癌农杆菌的dna序列、抗性基因等),尤其对于木本科植物白桦来说,个体发育的生命周期时间本身较长,虽然通过有性杂交技术可以去除这些外源基因,但所需周期会更长。目前,通过微粒轰击技术将cas9蛋白-grna核糖核蛋白复合物(rnp)导入受体细胞,可获得无选择标记基因、无t-dna插入的基因编辑植物,该技术的提出为快速创制白桦植物突变体提供了崭新思路。为了利用此技术进行白桦基因编辑研究,本发明以白桦glk基因为编辑靶基因,将体外纯化的cas9蛋白与glk-grna混合后的核糖核蛋白复合体(rnp),通过微粒轰击白桦愈伤组织,初步建立无t-dna插入基因编辑技术,为今后创制白桦突变体提供参考。
技术实现要素:
本发明的目的是提供一种靶向敲除glk基因的crispr/cas9系统及其应用,以解决上述现有技术存在的问题,通过设计、构建、筛选,最终提供一些基于crispr/cas9系统,同时能靶向白桦glk基因的高效grna及靶位点序列,并用其获得了无选择标记基因、无t-dna插入的基因编辑的白桦黄叶植株,该技术的提出为快速创制白桦植物突变体提供了崭新思路。
为实现上述目的,本发明提供了如下方案:
本发明提供一种敲除glk基因的grna,包括以下所述序列的任意一条:
grna-1:5′-gtcagctttgaggaacacaaagg-3′;
grna-2:5′-actacccagacttctccgatggg-3′;
grna-3:5′-gccgacttatccgttagcggcgg-3′;
grna-4:5′-ccgacttatccgttagcggcggt-3′。
本发明还提供一种grna位点靶向敲除glk基因的crispr/cas9系统,包括所述的grna-1、grna-2、grna-3以及grna-4中的任意一条dna序列。
本发明还提供根据所述的grna位点靶向敲除glk基因的crispr/cas9系统构建glk基因敲除的植物突变体的方法,包括以下步骤:以glk基因为编辑靶基因,将体外纯化的cas9蛋白与glk-grna混合后的核糖核蛋白复合体,通过微粒轰击植物体愈伤组织,转化后的愈伤组织经过继续培养和植株再生,获得无选择标记基因、无t-dna插入的基因编辑的植物突变体。
优选的是,所述植物愈伤组织来源于白桦。
本发明还提供一种根据所述的敲除glk基因的grna或所述的grna位点靶向敲除glk基因的crispr/cas9系统在构建glk基因敲除的植物突变体中的应用。
优选的是,所述植物突变体为白桦突变体。
本发明公开了以下技术效果:
本发明以白桦glk基因为编辑靶基因,将体外纯化的cas9蛋白与glk-grna混合后的核糖核蛋白复合体(rnp),通过微粒轰击白桦愈伤组织,初步建立无t-dna插入基因编辑技术。该技术改变了现有技术中直接采用crispr/cas9系统定点编辑时,宿主细胞仍然含有外源dna序列,以及利用有性杂交去除这些外源基因增加生育周期的缺陷等,经过该技术创建的物t-dna插入的编辑技术,获取了白桦黄叶植株并且能正常利用叶绿体生长,为丰富白桦品种以及快速创制白桦植物突变体提供了崭新思路。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
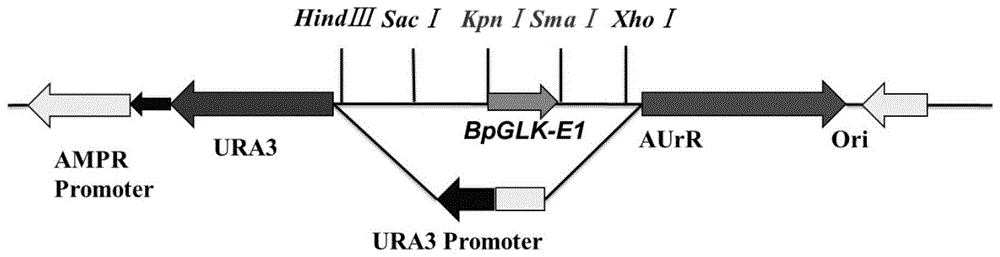
图1为pabai-glk-e1重组载体简图;
图2为pabai-glk-e1酶切电泳图谱;其中,m:dl15000;1-4pabai-glk-e1质粒;
图3为crisppr/cas9酶切靶点序列效率图;其中,m:dl10000;1-4:依次为由grna1-4与cas蛋白构建的grna-cas9蛋白复合物;5:阳性对照;6:空载(阴性对照);7:无cas9蛋白对照;8:无grna对照;
图4为愈伤组织不同培养阶段生长状态;其中,a为合子胚培养阶段;b和c为体式显微镜下的glkc愈伤组织;
图5为glk基因编辑株系测序比对图;
图6为体式显微镜下观察获得的白桦glk编辑植株;其中,a、b为glkc株系;c为对照白桦。
具体实施方式
现以实施例方式对本发明技术方案进行具体说明,但其不应该被认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。以下实施例中无特殊说明所使用的试剂或其他材料均能通过商业渠道获取。
以下实施例中所使用的白桦相关材料采集于东北林业大学白桦强化种子园。
实施例1bpglk基因靶位点的预测及grna活性检测
1、bpglk基因靶位点的预测以及grna序列设计
根据白桦bpglk(bpev01.c0167.g0013.m0001)的基因序列和编码区序列,利用crispr-direct网页(http://crispr.dbcls.jp/)筛选具有特异性靶点。即找到该基因的cds区序列中ngg(n为a,t,c或g),最好是agg,取ngg前面的20bp作为目标靶序列,以便提高基因敲除效率。根据靶点序列设计grna,具体见表1。
表1grna序列
2、grna的合成及活性检测
在grna序列前端加上t7启动子序列:taatacgactcactatag;在grna序列后端加上向导序列gtttaagagctatgctggaaacagcatagcaagtttaaataagg(茎环结构);利用表2grna合成用引物、表3grna的保守区段序列、以及表4pcr扩增体系、表5pcr扩增条件形成双链dna模板;随后进用nebt7快速高产rna合成试剂盒进行grna体外转录。
表2grna合成用引物序列
表3合成grna的保守区段序列
表4pcr反应体系
表5pcr反应条件
3、体外活性鉴定表达载体的构建
3.1将包含bpglk第一外显子序列的946bp序列(如seqidno:5所示,加下划线部分由前至后依次为grna-1、grna-2、grna-3,斜体部分为grna-4)构建到pabai载体上(注:长度过短会影响体外切割效率的验证,导致检测谱带距离太近,进而影响实验结果)。使用双酶切方法对质粒和bpglk-e1基因纯化产物进行酶切后连接并转化大肠杆菌。挑取单克隆做菌液pcr检测。将目标条带正确pabai-glk-e1重组质粒(如图1所示)送去测序,结果显示基因序列正确,可用于后续实验。
seqidno:5如下所示:
ctctccagtcttccttccgattcaccggaaaaagaaaaaaaaaaaaaaaaaaaaaaacaccttcaaacatataatttggtgggtgtttttttttttttggttacttcatgcagtagcaggaaattgcaccggagataatatatatatatattttggaattcatgcttgctgtgtcagctttgaggaacacaaaggatgaaaaccaaggagagttttcaatcggagtcaacgactacccagacttctccgatgggaatttgctcgacagcatcgatttcgacgatcttttcgtggatatcaacgacggagacgtgttgccggatttggaaatggacccggaaatgctcgccgacttatccgttagcggcggtgaggaatccgagatgtacacatccatgtctctcgaaaaattagacgatattactaattatacaaagaaagaagatcaggaagacaaagtttccggttccggttcgggctccggttccggttccggttcaagtcgaggagatgaaattgtcagcaaaagagatgaatctgttgtggtcaaaccacctccaaaacaagctgataataataaacccagaaaatcatcttcacactcaaagaattctcatggaaagcgcaaagtgaaggtaataataattaccatttgctttaaactctctctctctctctctctctttctcttatttgggtcttcataaacttcatgaggggcaaagaaatcacgggaatcatgggcactgacacaaatacacacagagtctgtgaagacccagaattgttttttttttaaaaaaaaaaatttggtgatattatttttgttcttgaattgtgtgtatataggtggattggaccccagaattgcacagaaggtttgtgcaagctgtggagcagctaggggtggataaggctgtgccttcaaggattttagagcttatgggaattgattgtctcacccgccacaacatagccagccaccttcag
3.2将得到的pabai-glk-e1重组质粒载体,利用bstei限制酶酶切,将获得的pabai-glk-e1重组质粒线性化后用于体外检测,稀释质粒dna浓度至约24.8ng/l,并在pcr扩增仪37℃温育3h,反应体系:pabai-glk-e1质粒20μl;bstei限制酶10μl;10×buffer20μl;ddh2o150μl;使用1%琼脂糖凝胶电泳检测质粒酶切结果。酶切产物电泳检测结果如图2所示,显示重组质粒载体在5000bp-7500bp处可见清晰酶切条带,大小与pabai-glk-e1质粒6000bp吻合。
3.3pabai-glk-e1质粒中靶位点的体外基因编辑实验准备如下表6所示的反应混合物以检测cas9核酸酶活性及合成的4个grna(grna-1、grna-2、grna-3、grna-4)能否定向切割靶序列。实验中对照组1设置无cas9蛋白,对照组2设置无grna、对照组3设置空质粒,充分混合反应体系后在37℃下孵育1h,然后立即将样品放入液氮。酶切产物用1%琼脂糖凝胶电泳检测,根据线性化的pabai-glk-e1质粒谱带情况,判断cas9核酸酶活性,检测grna的定性切割效率。
表6体外基因编辑反应体系
注:在无rnase的情况下进行
通过体外验证实验表明,cas9蛋白以及grna复合体具有裂解活性。能够将检测的4个bpglk靶位点在特定靶点进行切割,由图3可知4个靶点均在500bp-1000bp处检测到谱带,与我们设计的946bp目的基因靶点区域理论值符合。实验中对照组1无cas9蛋白,对照组2无grna、对照组3空质粒阴性对照则未见定点剪切谱带。体外bpglk靶位点检测表明,实验设计合成的4个grna均有活性。
实施例2基因枪介导的bpglk基因编辑
1、基因枪微粒制备(以下操作步骤按照每枪500μg的量可以轰击60枪)
(a)取30mg1.0μmgoldmicrocarriers加入eppendorf管。
(b)加入1ml70%酒精,充分涡旋,静置15min,1500rpm离心5min。
(c)弃上清,加入1ml无菌水,涡旋充分,1500rpm离心5min,进行3次重复。
(d)加入1ml的50%甘油,此时终浓度达到了60mg/ml,4℃保存。
2、crispr/cas9(rnp)的包裹(以下的量足够6枪,根据需要调整量)
(a)涡旋上述制备的goldmicrocarriers母液5min以打碎成团的微粒。需要注意的是,微粒很容易下沉,每一枪吸取的时候都要重新振荡重悬微粒。
(b)取50μlgoldmicrocarriers悬液于1.5mleppendorf管。
(c)依次加入crispr/cas9rnp复合物[在室温(25℃)下将5μlcas9蛋白和5μlgrna(grna体外验证已获得)混合,总体积为10μl]、50μl2.5mol/lcacl2、20μl0.1mol/l亚精胺(现用现配)。注意:边涡旋边加入,每加完一样,振荡2~3s。
(d)上述混好的样品,涡旋1min,冰上静置1min,重复3次。
(e)上静置10min,10000rpm离心10s,去上清。
(f)200μl无菌水冲洗沉淀2次,10000rpm离心10s,去上清。
(g)后用40μl无菌水重悬微粒。
3、基于基因枪法介导的bpglk基因编辑
采用pds-1000/he型基因枪操作,具体如下:
(a)转化前先把白桦愈伤组织(优选白桦合子胚接种到愈伤诱导培养基中置于暗处进行培养,温度条件为25℃,15d左右获得旺盛、松软、嫩白色的愈伤组织,面积约5角硬币大小)集中放在培养皿的培养基中心,直径2~3cm左右。在超净台上,75%酒精棉擦洗轰击室,同时消毒各配件,然后晾干。取8μldna包被的微粒点在载体膜中心,均匀分散开,避免堆积,把载体膜放到无水氯化钙的培养皿中干燥,其中氯化钙上铺一张无菌滤纸。
(b)打开真空泵和基因枪的电源开关进行基因轰击
打开氦气泵,逆时针旋转氦气总开关以打开氦气,此时钢瓶内部压力表应当显示钢瓶内部压力。顺时针旋转输出压力调节阀门,动作要慢,使输出压力表显示可裂膜的psi+200psi(优选轰击压力为1100psi,射程12cm的轰击方式)。
(c)拧下可裂膜挡盖,将可裂膜放在挡盖中央处(要放正,以防止泄漏空气),将挡盖旋上,用专用扳手加固。
(d)把微粒发射装置从轰击室拿出来,旋下盖子,加入阻挡网(阻挡网可重复利用,只要是同一个目的基因),把微粒载片安装在固定槽中(有微粒的一面朝下),旋上盖子,将微粒发生装置放置回轰击室。将样品培养皿放入轰击室的适当位置,关上轰击室门。
(e)取下微粒发生装置,卸下微粒载体膜和阻挡网。旋下可裂膜挡盖,清除可裂膜碎片等杂物。
(f)关掉氦气瓶的主阀门,按住fire开关,放掉气体加速管内的氦气压力,最后关闭一切电源。
注:亚精胺一般现用现配,如经常使用,可配成1mo/l的母液,分装于eppendorf管中,可于-20℃保存1-2个月;
4、转化后的白桦愈伤组织培养及植株再生
将转化后愈伤组织,28℃黑暗下培养过夜,然后从愈伤诱导培养基上转移到继代培养基上培养一周,如图4所示。一周后,将愈伤组织从继代培养基转移到分化培养基上,选择松软,分化良好的愈伤组织,弃去衰老褐化死亡的愈伤。每两周分化培养一次,培养至分化出嫩芽。分化期过后,将选择出的良好的嫩芽移至的继代培养基上,在28℃下培养,每两周继代培养一次。当分化的小植株长到1~2cm高时,转移到生根培养基继续培养。
其中,愈伤诱导培养基:wpm+2.0mg/l6-ba+0.2mg/lnaa+蔗糖20g/l+琼脂5.5g/l,暗培养(全遮光)15d-25d。继代培养基:将轰击后的愈伤组织转接于新鲜的培养基中(wpm+1.0mg/l6-ba+0.02mg/lnaa+0.5mg/lga3+卡那霉素+蔗糖20g/l+琼脂5.5g/l),暗培养(全遮光)3~5d。诱导及分化培养阶段:3~5d后将上述愈伤组织转接到诱导愈伤分化的培养基中,wpm+1.0mg/l6-ba+0.02mg/lnaa+0.5mg/lga3+蔗糖20g/l+琼脂5.5g/l)半遮光条件下培养15~25d。生根培养阶段:每隔25d一次培养基,转接3-4次后待愈伤组织分化成苗后,转接到生根培养基(wpm+0.4mg/liba+蔗糖200g/l+琼脂5.5g/l)中。其中,向导grna3获得了转bpglk基因编辑株系glkc。
以glkc株系的叶片总dna作为模板,用特异引物glk-grna-f:ctctccagtcttccttccgat,glk-grna-r:ctgaaggtggctggctatgt进行pcr检测,pcr反应体系见表7。随后进行pcr扩增,扩增程序为:94℃3min预变性,94℃30s变性,59℃30s退火,72℃延伸70s,35个循环,72℃7min,16℃保温。电泳结束后,用1%的琼脂糖凝胶对pcr产物进行检测。pcr产物在1%的琼脂糖凝胶进行电泳检测,通过snapgene软件将测序结果进行比对,如图5所示,我们发现1株glkc编辑株系均在调节叶色glk基因315-332处有18个碱基的删除,说明bpglk已经成功编辑了白桦基因组中。
表7glkc突变株检测pcr反应体系
对获得的glkc基因编辑株系继代苗叶色进行观察。如图6所示,结果发现,继代培养的glkc株系叶色呈现黄绿色,与野生型白桦呈现明显差异。
以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
序列表
<110>东北林业大学
<120>靶向敲除glk基因的crispr/cas9系统及其应用
<160>5
<170>siposequencelisting1.0
<210>1
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>1
gtcagctttgaggaacacaaagg23
<210>2
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>2
actacccagacttctccgatggg23
<210>3
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>3
gccgacttatccgttagcggcgg23
<210>4
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>4
ccgacttatccgttagcggcggt23
<210>5
<211>990
<212>dna
<213>人工序列(artificialsequence)
<400>5
ctctccagtcttccttccgattcaccggaaaaagaaaaaaaaaaaaaaaaaaaaaaacac60
cttcaaacatataatttggtgggtgtttttttttttttggttacttcatgcagtagcagg120
aaattgcaccggagataatatatatatatattttggaattcatgcttgctgtgtcagctt180
tgaggaacacaaaggatgaaaaccaaggagagttttcaatcggagtcaacgactacccag240
acttctccgatgggaatttgctcgacagcatcgatttcgacgatcttttcgtggatatca300
acgacggagacgtgttgccggatttggaaatggacccggaaatgctcgccgacttatccg360
ttagcggcggtgaggaatccgagatgtacacatccatgtctctcgaaaaattagacgata420
ttactaattatacaaagaaagaagatcaggaagacaaagtttccggttccggttcgggct480
ccggttccggttccggttcaagtcgaggagatgaaattgtcagcaaaagagatgaatctg540
ttgtggtcaaaccacctccaaaacaagctgataataataaacccagaaaatcatcttcac600
actcaaagaattctcatggaaagcgcaaagtgaaggtaataataattaccatttgcttta660
aactctctctctctctctctctctttctcttatttgggtcttcataaacttcatgagggg720
caaagaaatcacgggaatcatgggcactgacacaaatacacacagagtctgtgaagaccc780
agaattgttttttttttaaaaaaaaaaatttggtgatattatttttgttcttgaattgtg840
tgtatataggtggattggaccccagaattgcacagaaggtttgtgcaagctgtggagcag900
ctaggggtggataaggctgtgccttcaaggattttagagcttatgggaattgattgtctc960
acccgccacaacatagccagccaccttcag990
- 还没有人留言评论。精彩留言会获得点赞!