一种新型不对称催化亲核氟杂环化反应体系及其在手性非天然氨基酸模块合成中的应用的制作方法
本发明属于有机合成-不对称催化氟化技术领域,具体涉及一种新型不对称催化亲核氟杂环化反应体系及其应用。
背景技术:
在有机分子中引入一个或者多个氟原子(或含氟基团),能够极大改变该分子的物化性质。氟代分子往往比原分子表现出更强的亲脂性,更高的热稳定性和代谢稳定性,极性和分子间作用力减弱(petrone,d.a.;ye,j.;lautens,m.moderntransition-metal-catalyzedcarbon–halogenbondformation,chem.rev.2016,116,8003-8104)。含氟化合物在生物医药、材料科学和农业化学领域有着举足轻重的应用(champagne,p.a.;desroches,j.;hamel,j.d.;vandamme,m.;paquinj.f.monofluorinationoforganiccompounds:10yearsofinnovation.chem.rev.2015,115,9073-9174.)。但是天然产物中含氟有机化合物却极少(大约只有十余种结构简单的含氟有机分子),因此有机化合物的氟化,成为近年来的研究热点且热度持续不减(zhu,y.;han,j.l.;wang,j.d.;shibata,n.;sodeoka,m.;soloshonok,v.a.;coelho,j.a.s.;toste,f.d.modernapproachesforasymmetricconstructionofcarbon-fluorinequaternarystereogeniccenters:syntheticchallengesandpharmaceuticalneeds.chem.rev.2018,118,3887-3964.)。
鉴于不对称催化合成在有机化学中的重要应用以及手性化合物在药物化学领域的独特需求,使得不对称合成氟代分子(有机分子的手性氟化)成为有机氟化学中最引人注目,也是最具挑战且在诸多领域有巨大应用潜力的研究方向。由于氟原子的特殊性(如目前已知元素周期表中电负性最大的元素,氟原子的水合能高等),使得有机分子的手性氟(单氟或全氟)化的发展相对缓慢(ma,j.a.;cahard,d.asymmetricfluorination,trifluoromethylation,andperfluoroalkylationreactions.chem.rev.2004,104,6119-6146.)。
手性氟化学的发展,尤其是手性含氟化合物的催化合成,依赖于催化体系即氟试剂和催化剂体系的发展。就不对称催化氟化本身来说,其亲电氟化的发展远远成熟于亲核过程。当前,不对称催化氟化反应主要采用金属催化体系,但随着绿色化学和可持续化学发展的概念逐渐深入人心,金属催化体系由于在使用过程中存在诸如贵金属浪费、重金属毒性、需要复杂配体从而使使用成本增高等问题,日趋成为研究者们讨论的重要问题之一。因此,发展非金属参与的不对称催化氟化体系,具有重要的研究及应用意义。
近年来,出现了一批应用于不对称催化氟化反应的非金属催化体系,这些体系中用到的氟源通常包括如nfsi,selectfluor,et3n·hf,py·hf等,虽然这些非金属催化体系在手性氟化反应中成果丰硕,但是在制备或使用这些氟试剂或者有机催化剂过程中仍然存在制备复杂,原子利用率低,毒性大等问题。因此,发展新的不对称催化体系,从而通过手性氟化特别是不对称亲核氟化反应取得理想氟化产物,已成为该研究领域当前重要且富有意义的挑战。
一般来说,非天然氨基酸的合成方法主要以有机化学合成为主,少部分如酪氨酸类似结构的非天然氨基酸通过生物酶催化合成。就有机合成非天然氨基酸的方法来说,最直接的是对天然氨基酸进行化学修饰,可以得到单一手性构型的非天然氨基酸。但是该过程需要慎重设计合成路线,合理保护活性基团(氨基、羧基);另一种方法则是预先构建目标氨基酸侧链,然后与氨基酸手性中心前体化合物通过化学方法连接。该方法反应简便,合成步骤相对较少,但得到的往往是外消旋产物。迄今为止,不同构型的具有光学活性的非天然氨基酸的合成仍是一项具有挑战性的工作。为了克服上述难题,发明一种非金属催化的、能够得到手性功能化非天然氨基酸及其衍生物的方法,在生物医药合成、多肽药物合成、天然活性分子合成领域,都具有重要的研究及现实意义。
技术实现要素:
为了克服上述现有技术的不足,本发明提供了一种新型不对称催化亲核氟杂环化反应体系并将该反应体系应用于氟代噁嗪衍生物的制备中,该反应体系将手性高价碘催化剂和醚合三氟化硼组合,通过该反应体系对不饱和酰胺进行催化不对称亲核氟杂环化反应,可以生成手性氟代噁嗪衍生物,该手性氟代噁嗪衍生物经过水解后,可得到手性氟代氨基醇化合物或者手性非天然氨基酸。该反应体系具有反应条件温和,催化剂制备简单,无重金属污染和贵金属浪费,催化效率高,选择性好,对生产装置的腐蚀性低,且催化剂可以重复使用等优点。
为了实现上述目的,本发明所采用的技术方案是:
本发明提供一种新型不对称催化亲核氟杂环化反应体系在制备氟代噁嗪衍生物中的应用。
本发明还提供一种新型不对称催化氟杂环化反应体系,包括手性碘催化剂和醚合三氟化硼和不饱和酰胺。
优选地,所述手性碘催化剂包括但不限于如式ⅰ所示的具有中心对称结构的芳香碘化合物,或如式ⅱ所示的螺环手性碘化合物:
其中,r1=甲基、异丙基、苄基;r2=乙基、异丙基、苄基、叔丁基、薄荷醇基、金刚烷基等;r3=氢,乙基等。
进一步地,所述手性碘式ⅰ所示的中心对称的手性碘化合物,(标记为cic1),其中,r1=r2=苄基。
进一步地,所述手性碘催化剂可参考文献(banik,s.m.;medley,j.w.;jacobsen,e.n.catalytic,asymmetricdifluorinationofalkenestogeneratedifluoromethylatedstereocenters.science2016,353,51-54.)中的方法合成,具体合成流程如下所示:
优选地,所述醚合三氟化硼包括但不限于三氟化硼乙醚。
优选地,所述不饱和酰胺具有如下的结构式:
其中,ar1=苯基、4-氟苯基、4-甲氧基苯基、4-三氟甲基苯基、4-氟/氯/溴-苯基、或萘基;ar2=苯基、2或3或4位单取代基苯基、3,5-二甲氧基苯基、3,4-二甲氧基苯基、3,5-二叔丁基苯基,3,5-二三氟甲基苯基、2,3,4,5,6-五氟苯基、(苯并)-2-呋喃基、(苯并)-2-噻吩基、1-萘基、或2-萘基。
进一步地,底物不饱和酰胺可以通过简单的合成方法制备,具体合成流程如下所示:
具体地,底物不饱和酰胺的合成方法可根据参考文献(bardiasoltanzadeh,arvindjaganathan,richardj.staples,babakborhan,highlystereoselectiveintermolecularhaloetherificationandhaloesterificationofallylamides,angew.chem.int.ed.2015,54,9517-9522.)中的合成方法进行,具体的合成方法包括以下步骤(以ar1=ar2=苯基为例,标记为1a):
肉桂醇i(5mmol,1.0equiv.),邻苯二甲酰亚胺(5.5mmol,1.1equiv.)和三苯基膦(5.5mmol,1.1equiv.)溶解到四氢呋喃(thf)中(5ml/mmol),降温至0℃后,向其中缓慢加入偶氮二甲酸二异丙酯(diad,5.5mmol,1.1equiv.)。继续反应30-60分钟,tlc监测,当肉桂醇反应完全后,向其中缓慢加入水合肼(3.0equiv.),然后反应在室温下搅拌过夜。反应完毕后,向其中加入水稀释反应,然后加入3ml浓盐酸,继续搅拌30分钟。过滤,滤饼用5ml10%稀盐酸洗两次。收集水相,浓缩后得到桂皮胺盐酸盐ii,于40℃真空干燥48小时后直接投入下一步反应。
上述制备得到的桂皮胺盐酸盐(4mmol,1.0equiv.),三乙胺(20mmol,5.0equiv.)和催化量的4-二甲氨基吡啶(dmap,10mol%,0.1equiv.)加入到20ml干燥thf中,降至0℃后,向其中缓慢加入苯甲酰氯(6mmol,1.5equiv.)。滴加完毕后,升至室温继续反应。原料反应完全(tlc检测,大约3小时)后,加入1.0ml甲醇淬灭反应。然后加入20ml水,减压除去thf后,二氯甲烷(dcm)萃取(25ml×3次)。合并有机相,有机相使用20ml饱和食盐水洗涤后无水硫酸钠干燥。浓缩,柱层层析(洗脱液:pe/ea(v/v)=5:1)得到目标产物1a(白色固体,85%产率)。
其他底物的合成如无特殊说明,一概按照本领域常规方法进行。
具体地,所述底物不饱和酰胺的反应浓度范围为0.0125mol/l-0.2mol/l,最佳的反应浓度为0.025mol/l。
优选地,所述氟代噁嗪衍生物具有如下的结构式:
所得氟代噁嗪衍生物具有两个手性中心,同时具有高产率、高度化学选择和立体选择性,产品的光学活性高,可达到99%ee、>20:1dr和80%分离产率。
优选地,所述不饱和酰胺包括但不限于n-cinnamylbenzamide(n-肉桂基苯甲酰胺),n-cinnamyl-4-methoxybenzamide(n-肉桂基-4-甲氧基苯甲酰胺),n-cinnamyl-2-naphthamide(n-肉桂基-2-萘甲酰胺)。
优选地,所得到手性氟代苯并n,o-杂环化合物通过x射线单晶衍射确定的,其绝对构型是(s,s)型。
本发明还提供上述的一种新型不对称催化氟杂环化反应体系在不饱和酰胺的催化不对称亲核氟杂环化反应中的应用。
本发明又提供一种氟代噁嗪衍生物的制备方法,该制备方法通过催化不饱和的不对称亲核氟杂环化反应生成氟代化合物,即以上述的新型不对称催化亲核氟杂环化反应体系中的手性碘催化剂作为催化剂,醚合三氟化硼作为亲核氟试剂,对不饱和酰胺进行催化不对称亲核氟杂环化反应,经反应后处理得到具有高度化学选择性和立体选择性的氟代噁嗪衍生物。
本发明中,以手性碘催化剂(cic)和醚合三氟化硼组合构成催化体系,实现对不饱和酰胺的催化、不对称亲核氟杂环化反应;该过程中,手性碘试剂作为催化剂,醚合三氟化硼被首次应用于不对称亲核氟杂环化反应中;底物不饱和酰胺经过该催化体系,可以以高的化学选择性、高的分离产率(可达80%)、高的对映选择性(可达99%ee)和非对映选择性(可达>20:1dr)得到手性氟代噁嗪衍生物;该过程的化学选择性主要表现在:经过该催化体系,不饱和酰胺底物只生成单一手性氟代产物,而不会生成其他环化产物;本发明中,所用到的手性碘催化剂是参考相关文献直接合成,通过lc-ms,nmr表征后使用,并且在固定其他条件下,对催化剂进行了筛选以得到最佳催化剂。本发明的催化不对称亲核氟杂环化反应的化学选择性如以下的反应通式所示:
优选地,加入手性碘催化剂的量为5-20mol%(相对于反应底物不饱和酰胺),最佳的加入量为20mol%(相对于反应底物不饱和酰胺)。
优选地,加入亲核氟试剂(醚合三氟化硼)的量为反应底物(不饱和酰胺)摩尔量的10倍。
优选地,反应所用到的溶剂包括但不限于二氯甲烷、甲苯、氯仿、1,2-二氯乙烷(dce)、苯、氟苯、氯苯和三氟甲苯中的至少一种。
进一步地,反应所用到的溶剂为1,2-二氯乙烷(dce)。
优选地,反应过程中还需加入氧化剂。
进一步地,所加入的氧化剂为间氯过氧苯甲酸(m-cpba)。具体地,所加入的氧化剂的量为反应底物(不饱和酰胺)的0.8-1.5倍,最佳加入量为1.2倍。
优选地,反应的温度为室温(25℃)到-25℃,进一步地,反应温度为-25℃。
优选地,反应的时间为0.08-24h,进一步地,反应的时间为24h。
优选地,所述反应后处理是将反应后的混合体系倒入饱和碳酸氢钠溶液,然后用萃取柱进行分离,分离所用的洗脱液为石油醚(pe):乙酸乙酯(ea)=100:1-5:1。
优选地,反应完毕后,对于cic1催化剂可以经过简单的分离回收(柱分离回收),然后再重复使用。本发明的手性碘催化剂重复使用10次以上,仍然具有很好的催化活性和催化选择性。
作为本发明的一个优选实施例,以底物n-肉桂基苯甲酰胺(1a)为例,本发明的氟代噁嗪衍生物的制备方法具体包括以下步骤:
在15ml反应管中,加入1a(47.4mg,0.2mmol,1.0equiv.),催化剂cic1(28.5mg,20mol%)和8mldce,降温至-25℃并保持在该温度10分钟,然后向其中加入m-cpba(0.24mmol,1.2equiv.)并缓慢滴入bf3·et2o(2mmol,10.0equiv.)。滴加完毕后,旋紧反应管塞子,继续在-25℃搅拌48小时。反应完毕后,将反应液倾入10ml饱和碳酸氢钠溶液,于分液漏斗中震荡静置分层,有机相继续使用10ml饱和碳酸氢钠溶液洗涤。有机相使用无水硫酸钠干燥后,碱性氧化铝过柱(洗脱液:pe/ea(v/v)=50:1,0.5%et3n),得到白色固体(产率70%,86%ee,>20:1dr)。
其他不饱和酰胺底物的不对称氟杂环化反应,如无特殊说明,均按照以上操作条件进行。
本发明再提供一种氟代手性氨基醇化合物和手性非天然氨基酸模块合成的方法。氨基醇尤其是氨基醇化合物是重要的合成中间体,在天然、生物活性分子,药物分子的合成中都有着重要的应用。本发明中得到的氟代噁嗪衍生物经水解后,可以得到手性氟代氨基醇化合物,该化合物经氧化后可得到手性非天然氨基酸,扩展了手性氨基酸及其衍生物的合成方法,得到的产品可用于药物分子、活性结构和多肽药物的合成等。氟代噁嗪衍生物经水解-氧化-水解反应后,得到芳基、杂环取代的手性非天然氨基酸,本发明的方法依照如下反应通式进行:
第一步水解反应(1):
优选地,反应过程需要用到碱溶液,该碱溶液包括碳酸钠水溶液、氢氧化钠水溶液、氢氧化钾水溶液中的至少一种。进一步地,用到的是naoh水溶液(2当量,10%~50%),其中,以20%的浓度最佳。
优选地,反应所用到的溶剂包括但不限于水,四氢呋喃、甲醇和乙醇中的至少一种。进一步地,反应所用到的溶剂为水和四氢呋喃(体积比为1:1)。
优选地,反应浓度为0.25-2mol/l。进一步地,反应浓度为0.5mol/l。
优选地,反应的温度为室温(25℃)到80℃。进一步地,反应温度为40℃。
优选地,反应的时间为1-24h。进一步地,反应的时间为8h。
优选地,反应结束后进行处理,处理过程为将反应后的混合体系倒入饱和碳酸氢钠溶液,然后用萃取柱进行分离,分离所用的洗脱液为石油醚(pe):乙酸乙酯(ea)=100:1-3:1。
第二步氧化反应(2):
优选地,氧化反应过程需加入氧化剂,加入的氧化剂为高价碘氧化剂如戴斯-马丁试剂,ibx试剂。进一步地,加入的氧化剂为ibx试剂。
优选地,加入氧化剂的量为0.8-2.5当量(相对于反应底物手性氨基醇),最佳的加入量为1.5当量(相对于反应底物手性氨基醇)。
优选地,反应所用到的溶剂包括但不限于二氯甲烷、甲苯、氯仿、1,2-二氯乙烷(dce)和氯苯中的至少一种。进一步地,反应所用到的溶剂为二氯甲烷(dcm)。
优选地,反应浓度为0.1-2mol/l。进一步地,反应浓度为0.1mol/l。
优选地,反应的温度为室温(25℃)到0℃。进一步地,反应温度为0℃。
优选地,反应的时间为1-12h。进一步地,反应的时间为6h。
优选地,反应结束后进行处理,处理过程为将反应后的混合体系倒入稀硫代硫酸钠水溶液中,然后用萃取柱进行分离,分离所用的洗脱液为石油醚(pe):乙酸乙酯(ea)=100:1-1:1。
第三步水解反应(3):
优选地,反应过程需要用到碱溶液,该碱溶液包括碳酸钠水溶液、氢氧化钠水溶液、氢氧化钾水溶液中的至少一种。
进一步地,用到的是naoh水溶液(0.5-6mol/l),其中,以1mol/l的浓度最佳。
优选地,反应所用到的溶剂包括但不限于水,四氢呋喃、甲醇和乙醇中的至少一种。进一步地,反应所用到的溶剂为甲醇(甲醇和氢氧化钠水溶液的体积比为1:1)。
优选地,反应浓度为0.25-2mol/l。进一步地,反应浓度为0.5mol/l。
优选地,反应的温度为室温(25℃)到80℃。进一步地,反应温度为80℃。
优选地,反应的时间为1-24h。进一步地,反应的时间为8h。
优选地,反应结束后进行处理,处理过程为向反应后的混合体系中加入1mol/l的盐酸调节ph至5~6,然后将混合液倒入饱和碳酸钠水溶液中,搅拌30分钟后,然后用萃取柱进行分离,分离所用的洗脱液为石油醚(pe):乙酸乙酯(ea)=100:1-1:2。
作为本发明的一个优选实施例,以产物1b的水解、氧化反应为例,本发明的手性非天然氨基酸的制备方法具体包括以下步骤:
在15ml反应管中,加入1b(510mg,1.0mmol,1.0equiv.)和四氢呋喃(2ml),降温至0℃后向其中加入2mlnaoh溶液(1mol/l),滴加完毕后,室温搅拌6小时。反应完毕后,将反应液倾入10ml饱和碳酸氢钠溶液,于分液漏斗中震荡静置分层,有机相继续使用10ml饱和碳酸氢钠溶液洗涤。有机相使用无水硫酸钠干燥、除去溶剂后直接投入下一步。上述产品溶解到10ml二氯甲烷中,降温至0℃后,向其中缓慢加入市售ibx(1.5mmol),继续搅拌6小时后,加入稀硫代硫酸钠淬灭反应。加水、二氯甲烷洗涤,有机相经硫酸钠干燥、除去溶剂后柱分离(洗脱液:石油醚(pe):乙酸乙酯(ea)=100:1-1:1)得到白色产物,两步产率85%。
上述得到的酰胺-羧酸产物(1mmol)和2ml甲醇加入到25ml圆底烧瓶中,降温至0℃后,加入2mlnaoh溶液(2mol/l),80℃回流8小时后,降至室温,向其中缓慢加入1n盐酸,调节ph至5~6。随后将混合液倒入10ml饱和碳酸钠溶液中,搅拌30分钟。乙酸乙酯萃取三次。合并有机相,有机相经无水硫酸钠干燥后柱分离(分离所用的洗脱液为石油醚(pe):乙酸乙酯(ea)=100:1-1:2。)得到白色产品,90%产率。
其他氟代噁嗪衍生物的水解-氧化-水解反应,如无特殊说明,均按照以上操作条件进行。
与现有技术相比,本发明的有益效果是:
本发明提供一种新型不对称亲核催化氟杂环化反应体系、该反应体系在制备氟代噁嗪衍生物中的应用以及氟代噁嗪衍生物的制备方法,该反应体系包括手性碘催化剂和醚合三氟化硼和不饱和酰胺,该反应体系,首次使用廉价易得的醚合三氟化硼作为亲核氟试剂,降低了生产成本,拓宽了催化不对称亲核氟杂环化反应的催化体系和反应类型;同时,该反应体系无重金属污染和贵金属浪费,环境友好,反应条件温和,化学选择性和立体选择性高,后处理简单,对生产装置的腐蚀性低。
将上述反应体系应用于不饱和酰胺的催化不对称亲核氟杂环化反应中,以反应体系中的手性碘催化剂作为催化剂,醚合三氟化硼作为亲核氟试剂,对不饱和酰胺进行催化不对称亲核氟杂环化反应,经反应后处理得到具有高度化学选择性和立体选择性的氟代噁嗪衍生物。与金属催化的不对称氟杂环化反应体系相比,本发明的催化反应具有以下优势:醚合三氟化硼作为氟试剂,廉价易得、稳定性好、毒性低,是目前市面最便宜的氟试剂;反应条件温和;手性碘催化剂制备简单,不需要复杂配体;环境友好,无重金属污染和贵金属浪费;得到的产物是具有两个手性中心的氟代噁嗪衍生物活性分子结构单元;反应的化学选择性(只得到单一氟代产物)和立体选择性好(可达到>99%ee和>20:1dr)。此外,对于手性碘催化剂,反应完毕后可以经过简单的柱层析分离回收,实现了催化剂的重复使用,符合绿色可持续化学发展的理念和要求,进一步降低了生产成本,节约了资源。
将上述得到的氟代噁嗪衍生物经过相应的化学转化后,可得到氟代手性氨基醇或手性非天然氨基酸化合物,具有重要的应用价值。该转化过程无金属参与、环境友好,产率高,转化过程光学活性保持良好。这对于功能化手性氨基醇化合物、手性氨基酸特别是非天然氨基酸的化学合成,提供了新的转化思路,在多肽合成、活性分子合成领域,具有重要研究及应用价值。
附图说明
图1为本发明实施例2得到的产物1b的消旋体和手性产物的hplc图谱;
图2为本发明实施例3得到的产物2b的消旋体和手性产物的hplc图谱;
图3为本发明实施例4得到的产物3b的消旋体和手性产物的hplc图谱;
图4为本发明实施例3得到的产物2b的x射线单晶衍射图;
图5为本发明实施例6得到的产物2b的x射线单晶衍射图。
具体实施方式
下面对本发明的具体实施方式作进一步说明。在此需要说明的是,对于这些实施方式的说明用于帮助理解本发明,但并不构成对本发明的限定。此外,下面所描述的本发明各个实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互组合。
下述实施例中的实验方法,如无特殊说明,均为常规方法,下述实施例中所用的试验材料,如无特殊说明,均为可通过常规的商业途径购买得到的。
实验材料和仪器:
低温反应仪,hplc,lc-ms,nmr,肉桂醇,l-苯丙氨酸,间氯过氧苯甲酸,超干四氢呋喃(thf),超干二氯甲烷(dcm),醚合三氟化硼(bf3·et2o),4-二甲氨基吡啶(dmap)苯甲酸,对甲氧基苯甲酸,对乙氧基苯甲酸,3,5-二叔丁基苯甲酸,呋喃-2-甲酸,苯并呋喃-2-甲酸,噻吩-2-甲酸,苯并噻吩-2-甲酸,1,2-二氯乙烷(dce),nacl,nahco3。
如无特殊说明:1)所有试剂均是市售分析纯商品;2)所有市售商品直接使用,未经进一步纯化处理;3)原料的柱层层析分离用到的硅胶均为200-300目。
实施例1新型不对称催化亲核氟杂环化反应体系的建立
该新型不对称催化亲核氟杂环化反应体系包括手性碘催化剂cic1和三氟化硼乙醚(bf3·et2o)。
其中,中心对称的手性碘催化剂cic1的制备方法如下:
根据参考文献(banik,s.m.;medley,j.w.;jacobsen,e.n.catalytic,asymmetricdifluorinationofalkenestogeneratedifluoromethylatedstereocenters.science2016,353,51-54)中的方法合成。
核磁表征:1hnmr(500mhz,cdcl3)δnmr(500mhz,cdcl,j.w.;jacobsen,e.n.catalytic,asymmetricdifluorinationofalkenestogeneratedifluoromethylatedstereocenters.science2016,353,51-54氟试剂,廉价易得、稳定性好、毒性低,是目前市面最便宜的氟试剂;反应条件温和;手性碘催化剂制备简单,不需要复杂配体;环境友好,无重金属污染和贵金属浪费;得到的产物是具有两个手性中心的氟代噁嗪衍0hz,2h);13cnmr(126mhz,cdcl3)nmr70.3,158.0,135.9,135.1,130.0,129.4,128.5,128.4,128.3(2c),127.1,106.1,79.6,78.8,67.0,39.1.
实施例2n-肉桂基苯甲酰胺(1a)的催化不对称亲核氟杂环化反应生成手性氟代噁嗪衍生物
本实施例的催化不对称亲核氟杂环化反应按照以下反应式进行:
具体操作方法为:
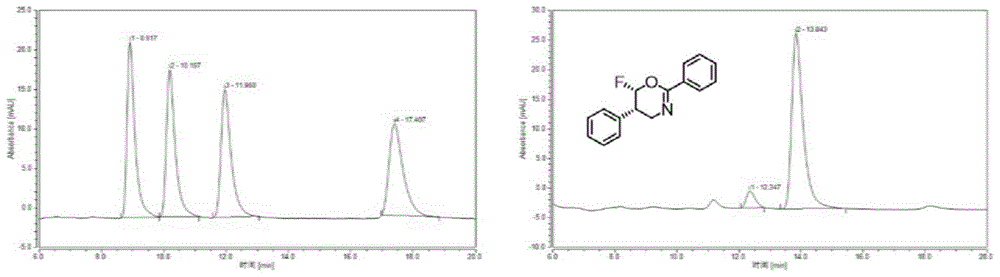
(1)在15ml反应管中,加入1a(47.4mg,0.2mmol,1.0equiv.),催化剂cic1(20mol%)和8mldce,降温至-25℃并保持在该温度10分钟,然后向其中加入m-cpba(0.24mmol,1.2equiv.)并缓慢滴入bf3·et2o(2mmol,10.0equiv.)。滴加完毕后,旋紧反应管塞子,继续在-25℃搅拌48小时。反应完毕后,将反应液倾入10ml饱和碳酸氢钠溶液,于分液漏斗中震荡静置分层,有机相继续使用10ml饱和碳酸氢钠溶液洗涤。有机相使用无水硫酸钠干燥后,碱性氧化铝过柱(洗脱液:pe/ea(v/v)=50:1,0.5%et3n),得到白色固体产物1b(产率70%,86%ee,>20:1dr)。
(2)产物1b的表征(lc-ms,hplc,nmr)
hplc:经过条件筛选,对得到的产物1b进行hplc测试,具体表征结果如下:
产物1b的hplc测试结果是:86%ee;
测试的条件为:色谱柱:phenomenexcellulose-1;洗脱液:i-proh/hexanes(v/v)=95:5,1.0ml/min;tr(major)=13.843min,tr(minor)=12.347min。消旋体和手性产物的hplc图谱如图1所示,其中,左图为消旋体的hplc出峰,右图为手性催化条件下得到的具有对映选择性和非对映的氟代产物,且右图两个出峰的积分面积相减可以看出对映体过量为86%,即86%ee;19fnmr可以看出,非对映体选择性为>20:1dr。
核磁、高分辨质谱表征:
1hnmr(400mhz,cdcl3)δ7.99-7.97(d,j=8.0hz,2h),7.45-7.33(m,8h),6.17-6.02(dd,j=3.2,56.8hz,1h),4.05-3.97(m,1h),3.87-3.80(m,1h),3.30-3.18(dddd,j=1.2,6.0,7.2,29.6hz,1h);13cnmr(126mhz,cdcl3)δ151.5,136.1,132.3,131.0,128.9,128.2,127.9,105.0(d,j=228.7hz),43.5(d,j=5.0hz),41.7(d,j=21.3hz);19fnmr(376mhz,cdcl3)δ117.2(s,1f),-132.0(s,25f).hrms(esi-tof)calc’dforc16h14fno[m+h]+:256.1132;found256.1130.
(3)催化剂cic1的回收再使用:催化剂经柱分离回收(石油醚/乙酸乙酯=10:1柱分离)后,再次按照本实施例反应条件投入催化循环,实验结果表明,使用10次以后,催化剂仍然具有一定的催化活性。
实施例3n-肉桂基-4-甲氧基苯甲酰胺(2a)的催化不对称亲核氟杂环化反应生成手性氟代噁嗪衍生物
本实施例的催化不对称亲核氟杂环化反应按照下列反应通式进行:
具体操作方法为:
(1)在15ml反应管中加入2a(53.5mg,0.2mmol,1.0equiv.),催化剂cic1(20mol%)和8mldce,降温至-25℃并保持在该温度10分钟,然后向其中加入间氯过氧苯甲酸(m-cpba,0.24mmol,1.2equiv.),并缓慢滴入bf3·et2o(2mmol,10.0equiv.)。滴加完毕后,旋紧反应管塞子,继续在-25℃搅拌24小时。反应完毕后,将反应液倾入10ml饱和碳酸氢钠溶液中,于分液漏斗中震荡静置分层,有机相继续使用10ml饱和碳酸氢钠溶液洗涤。有机相使用无水硫酸钠干燥后,快速柱层析分离(洗脱液:pe/ea(v/v)=20:1,0.5%et3n),得到白色固体产物2b(产率75%,99%ee,>20:1dr)。
(2)产物2b的表征(hplc,nmr,hrms)
hplc:经过条件筛选,对得到产物2b进行hplc测试,具体表征结果如下:
产物2b的hplc测试结果是:99%ee;
测试的条件是:色谱柱:phenomenexluxamylose-1;洗脱液:i-proh/hexanes(v/v)=90:10,1.0ml/min;tr(major)=12.703min,tr(minor)=9.537min。消旋体和手性产物的hplc图谱如图2所示,其中,上图为消旋体的hplc出峰,下图为手性催化条件下得到的具有对映选择性的氟代产物,且下图两个出峰的积分面积相减可以看出对映体过量为99%,即99%ee;19fnmr表明非对映体结果为>20:1dr。
核磁、高分辨质谱表征:
1hnmr(500mhz,cdcl3)57.93-7.92(d,j=9.0hz,2h),7.39-7.33(m,4h),7.27-7.26(d,j=9.0hz,2h),6.92-6.90(d,j=8.5hz,2h)6.14-6.01(dd,j=3.0,57.0hz,1h),4.02-3.96(m,1h),3.85-3.78(m,4h),3.27-3.18(m,1h);13cnmr(126mhz,cdcl3)nmr(.9,151.2,136.3,128.9,128.8,128.5,127.9,124.8,113.5,105.0(d,j=228.7hz),55.4,43.4(d,j=4.6hz),41.7(d,j=21.5hz);19fnmr(471mhz,cdcl3)n-117.2(s,1f),-132.0(s,26f).hrms(esi-tof)calc’dforc17h16fno2[m+h]+:286.1238;found286.1237.
(3)催化剂的回收再使用:同实施例2。
(4)产物2b的x射线单晶衍射图如图4所示,由图4中可以看出产物分子的立体构型是(5s,6s)。
实施例4n-肉桂基2-萘甲酰胺(3a)的催化不对称亲核氟杂环化反应生成手性氟代噁嗪衍生物
本实施例的催化不对称亲核氟杂环化反应按照下列反应通式进行:
具体操作方法为:
(1)在15ml反应管中,加入3a(57.5mg,0.2mmol,1.0equiv.),催化剂cic1(20mol%)和8mldce,降温至-25℃并保持在该温度10分钟,然后向其中加入m-cpba(0.24mmol,1.2equiv.)并缓慢滴入bf3·et2o(2mmol,10.0equiv.)。滴加完毕后,旋紧反应管塞子,继续在-25℃搅拌48小时。反应完毕后,将反应液倾入10ml饱和碳酸氢钠溶液,于分液漏斗中震荡静置分层,有机相继续使用10ml饱和碳酸氢钠溶液洗涤。有机相使用无水硫酸钠干燥后,碱性氧化铝过柱(洗脱液:pe/ea(v/v)=50:1,0.5%et3n),得到白色固体产物3b(产率77%,87%ee,>20:1dr)。
(2)产物3b的表征(lc-ms,hplc,nmr)
hplc:经过条件筛选,对得到的产物3b进行hplc测试,具体表征结果如下:
产物3b的hplc测试结果是:87%ee;
测试的条件为:色谱柱:ic,洗脱液:i-proh/hexanes(v/v)=98:2,1.0ml/min;tr(major)=15.657min,tr(minor)=14.203min。消旋体和手性产物的hplc图谱如图3所示,其中,上图为消旋体的hplc出峰,下图为手性催化条件下得到的具有对映选择性和非对映的氟代产物,且下图两个出峰的积分面积相减可以看出对映体过量为87%,即87%ee。19fnmr可以看出,非对映体选择性为>20:1dr。
核磁、高分辨质谱表征:
1hnmr(400mhz,cdcl3)n8.48(s,1h),8.10-8.07(d,j=8.8hz,1h),7.93-7.91(d,j=8.8hz,1h),7.88-7.86(d,j=8.8hz,2h),7.54-7.49(m,2h),7.41-7.35(m,5h),6.24-6.09(dd,j=3.6,56.8hz,1h),4.11-4.03(m,1h),3.93-3.86(m,1h),3.36-3.24(dddd,j=1.2,5.6,13.2,29.6hz,1h);13cnmr(126mhz,cdcl3)nmr1.6,136.1,134.7,132.8,129.6,129.0,128.9,128.5,128.5,128.0,127.7,127.6,127.4,126.4,124.1,106.0-104.2(d,j=228.7hz),43.6(d,j=5.2hz),41.9-41.7(d,j=21.4hz);19fnmr(471mhz,cdcl3)δ-117.1(s,1f),-131.9(s,30f).hrms(esi-tof)calc’dforc20h16fno[m+h]+:306.1289;found306.1281.
(3)催化剂cic1的回收再使用:催化剂经柱分离回收(石油醚/乙酸乙酯=10:1柱分离)后,再次按照本实施例反应条件投入催化循环,实验结果表明,使用10次以后,催化剂仍然具有一定的催化活性。
实施例5新型不对称催化亲核氟杂环化反应体系的建立
该新型不对称催化亲核氟杂环化反应体系包括手性碘催化剂cic2和三氟化硼乙醚(bf3·et2o)。
其中,中心对称的手性碘催化剂cic2的制备方法如下:
根据参考文献(dohi,t.,etal.,am.chem.soc.2013,135(11),4558-4566;dohi,t.,etal.,angew.chem.int.ed.2008,47,3787-3790)中的方法合成。
核磁表征:1hnmr(400mhz,cdcl3)δnmr(400mhz,j=8.0hz,2h),7.08-7.26(d,j=8.0hz,2h),3.06-3.02(m,4h),2.77-2.70(m,4h),2.49-2.41(m,2h),2.24-2.18(m,2h),1.19-1.15(t,j=7.5hz,6h);13cnmr(101mhz,cdcl3)nmr(101mhz,cdcl-3.02(m,4h),2.77-2.70(m,4h),2.49-4.4,30.1,15.1.
实施例6n-肉桂基2-呋喃甲酰胺(4a)的催化不对称亲核氟杂环化反应生成手性氟代噁嗪衍生物
本实施例的催化不对称亲核氟杂环化反应按照下列反应通式进行:
具体操作方法为:
(1)在15ml反应管中,加入4a(45.4mg,0.2mmol,1.0equiv.),催化剂cic2(10mol%)和8mldce,降温至-25℃并保持在该温度10分钟,然后向其中加入m-cpba(0.24mmol,1.2equiv.)并缓慢滴入bf3·et2o(2mmol,10.0equiv.)。滴加完毕后,旋紧反应管塞子,继续在-25℃搅拌48小时。反应完毕后,将反应液倾入10ml饱和碳酸氢钠溶液,于分液漏斗中震荡静置分层,有机相继续使用10ml饱和碳酸氢钠溶液洗涤。有机相使用无水硫酸钠干燥后,碱性氧化铝过柱(洗脱液:pe/ea(v/v)=50:1,0.5%et3n),得到白色固体产物4b(产率75%,88%ee,>20:1dr)。
(2)产物4b的表征(lc-ms,hplc,nmr)
hplc:经过条件筛选,对得到的产物4b进行hplc测试,具体表征结果如下:
产物4b的hplc测试结果是:88%ee;
测试的条件为:色谱柱:phenomenexluxamylose-1,洗脱液:i-proh/hexanes(v/v)=98:2,1.0ml/min;tr(major)=25.273min,tr(minor)=23.630min。消旋体和手性产物的hplc图谱如图5所示,其中,左图为消旋体的hplc出峰,右图为手性催化条件下得到的具有对映选择性和非对映的氟代产物,且右图两个出峰的积分面积相减可以看出对映体过量为887%,即88%ee。19fnmr可以看出,非对映体选择性为>20:1dr。
核磁、高分辨质谱表征:
1hnmr(400mhz,cdcl3)δ7.46-7.44(m,1h),7.31-7.26(m,5h),6.86-6.85(d,j=3.4hz,1h),6.42-6.40(m,1h),6.03-5.89(dd,j=3.2,56.4hz,1h),3.99-3.88(m,1h),3.80-3.73(m,1h),3.24-3.11(dddd,j=1.2,6.0,13.2,30.0hz,1h);13cnmr(101mhz,cdcl3)nmr(101mhz,cdcl0.0hz,.88(m,1h),3.80-3.73(m,1h),3.24-3.11(dddd,j=230.6hz),43.1(d,j=4.9hz),41.7,41.5(d,j=21.3hz);19fnmr(376mhz,cdcl3)δ-117.6(s,1f),-132.7(s,10f).hrms(esi-tof)calc’dforc14h12fno2[m+h]+:246.0925;found246.0927.
(3)催化剂cic1的回收再使用:催化剂经柱分离回收(石油醚/乙酸乙酯=10:1柱分离)后,再次按照本实施例反应条件投入催化循环,实验结果表明,使用10次以后,催化剂仍然具有一定的催化活性。
实施例7氟代噁嗪衍生物1b的水解-氧化-水解反应生成手性非天然氨基酸
选用实施例2的反应产物1b进行反应,本实施例的水解-氧化-水解反应依照下列反应通式进行:
(1)氟代手性氨基醇1c的合成
在15ml反应管中,加入1b(510mg,1.0mmol,1.0equiv.)和四氢呋喃(2ml),降温至0℃后向其中加入2mlnaoh溶液(1mol/l),滴加完毕后,室温搅拌6小时。反应完毕后,将反应液倾入10ml饱和碳酸氢钠溶液,于分液漏斗中震荡静置分层,有机相继续使用10ml饱和碳酸氢钠溶液洗涤。有机相使用无水硫酸钠干燥、除去溶剂后直接投入下一步。
(2)手性甲酰氨羧酸酸1d的合成
将步骤(1)中得到的产品1c溶解到10ml二氯甲烷中,降温至0℃后,向其中缓慢加入市售ibx(1.5mmol),继续搅拌6小时后,加入稀硫代硫酸钠淬灭反应。加水、二氯甲烷洗涤,有机相经硫酸钠干燥、除去溶剂后柱分离(洗脱液:石油醚(pe):乙酸乙酯(ea)=100:1-1:1)得到白色产物,两步产率85%。
(3)手性非天然氨基酸1e的合成
将步骤(2)中得到的酰胺-羧酸产物1d(1mmol)和2ml甲醇加入到25ml圆底烧瓶中,降温至0℃后,加入2mlnaoh溶液(2mol/l),80℃回流8小时后,降至室温,向其中缓慢加入1n盐酸,调节ph至5~6。随后将混合液倒入10ml饱和碳酸钠溶液中,搅拌30分钟。乙酸乙酯萃取三次。合并有机相,有机相经无水硫酸钠干燥后柱分离(分离所用的洗脱液为石油醚(pe):乙酸乙酯(ea)=100:1-1:2)得到白色产品1e,产率90%。
(4)产物1e的表征
1hnmr(500mhz,meod)δ12.72(b,1h),7.35-7.29(m,5h),3.62-3.59(dd,j=7.0hz,1h),3.09-3.34(m,2h);13cnmr(500mhz,meod),δ178.1,135.4,129.2,127.3,48.5,43.2.
实施例8氟代噁嗪衍生物5b的水解-氧化-水解反应生成手性非天然氨基酸
本实施例的水解-氧化-水解反应依照下列反应通式进行:
(1)手性氟代噁嗪衍生物5b的合成
参照实施例2的一般方法,由(e)-n-(3-(4-氟苯基)烯丙基苯并噻吩2-甲酰胺)(底物5a)经不对称催化氟杂环化反应得到手性氟代噁嗪衍生物5b,白色固体,产率82%。
(2)氟代手性氨基醇5c的合成
在15ml反应管中,加入5b(658mg,1.0mmol,1.0equiv.)和四氢呋喃(2ml),降温至0℃后向其中加入2mlnaoh溶液(1mol/l),滴加完毕后,室温搅拌6小时。反应完毕后,将反应液倾入10ml饱和碳酸氢钠溶液,于分液漏斗中震荡静置分层,有机相继续使用10ml饱和碳酸氢钠溶液洗涤。有机相使用无水硫酸钠干燥、除去溶剂后直接投入下一步。
(3)手性甲酰氨羧酸酸5d的合成
步骤(2)中得到的产品5c溶解到10ml二氯甲烷中,降温至0℃后,向其中缓慢加入市售ibx(1.5mmol),继续搅拌6小时后,加入稀硫代硫酸钠淬灭反应。加水、二氯甲烷洗涤,有机相经硫酸钠干燥、除去溶剂后柱分离(洗脱液:石油醚(pe):乙酸乙酯(ea)=100:1-1:1)得到白色产物,两步产率82%。
(4)手性非天然氨基酸5e的合成
步骤(3)中得到的酰胺-羧酸产物5d(1mmol)和2ml甲醇加入到25ml圆底烧瓶中,降温至0℃后,加入2mlnaoh溶液(2mol/l),80℃回流8小时后,降至室温,向其中缓慢加入1n盐酸,调节ph至5~6。随后将混合液倒入10ml饱和碳酸钠溶液中,搅拌30分钟。乙酸乙酯萃取三次。合并有机相,有机相经无水硫酸钠干燥后柱分离(分离所用的洗脱液为石油醚(pe):乙酸乙酯(ea)=100:1-1:2)得到白色产品5e,产率89%。
(5)产物5e的表征
1hnmr(500mhz,meod)δ12.52(b,1h),7.42-7.40(d,j=8.5hz,5h),7.16-7.13(dd,j=8.0hz,2h),3.83-3.79(dd,j=7.0hz,1h),3.12-3.35(m,2h);13cnmr(500mhz,meod),δ177.3,161.7(d,j=251.0hz),131.3,131.0,115.6(d,j=5.5hz),48.3,43.9.
实施例9氟代噁嗪衍生物6b的水解-氧化-水解反应生成手性非天然氨基酸
本实施例的水解-氧化-水解反应依照下列反应通式进行:
(1)手性氟代噁嗪衍生物6b的合成
参照实施例2的一般方法,由(e)-n-(3-(4-甲氧基苯基)烯丙基苯并噻吩2-甲酰胺)(底物6a)经不对称催化氟杂环化反应得到手性氟代噁嗪衍生物5b,白色固体,产率85%。
(2)氟代手性氨基醇6c的合成
在15ml反应管中,加入6b(682mg,1.0mmol,1.0equiv.)和四氢呋喃(2ml),降温至0℃后向其中加入2mlnaoh溶液(1mol/l),滴加完毕后,室温搅拌6小时。反应完毕后,将反应液倾入10ml饱和碳酸氢钠溶液,于分液漏斗中震荡静置分层,有机相继续使用10ml饱和碳酸氢钠溶液洗涤。有机相使用无水硫酸钠干燥、除去溶剂后直接投入下一步。
(3)手性甲酰氨羧酸酸6d的合成
步骤(2)中得到的产品6c溶解到10ml二氯甲烷中,降温至0℃后,向其中缓慢加入市售ibx(1.5mmol),继续搅拌6小时后,加入稀硫代硫酸钠淬灭反应。加水、二氯甲烷洗涤,有机相经硫酸钠干燥、除去溶剂后柱分离(洗脱液:石油醚(pe):乙酸乙酯(ea)=100:1-1:1)得到白色产物,两步产率88%。
(4)手性非天然氨基酸6e的合成
步骤(3)中得到的酰胺-羧酸产物6d(1mmol)和2ml甲醇加入到25ml圆底烧瓶中,降温至0℃后,加入2mlnaoh溶液(2mol/l),80℃回流8小时后,降至室温,向其中缓慢加入1n盐酸,调节ph至5~6。随后将混合液倒入10ml饱和碳酸钠溶液中,搅拌30分钟。乙酸乙酯萃取三次。合并有机相,有机相经无水硫酸钠干燥后柱分离(分离所用的洗脱液为石油醚(pe):乙酸乙酯(ea)=100:1-1:2)得到白色产品6e,产率91%。
(5)产物6e的表征
1hnmr(500mhz,meod)δ12.72(b,1h),7.22-7.20(d,j=8.5hz,2h),6.86-6.83(d,j=8.0hz,2h),3.80(s,3h),3.60-3.57(dd,j=7.0hz,1h),3.10-3.33(m,2h);13cnmr(500mhz,meod),δ177.3,159.5,130.7,128.4,114.8,55.7,47.8,44.0.
以上对本发明的实施方式作了详细说明,但本发明不限于所描述的实施方式。对于本领域的技术人员而言,在不脱离本发明原理和精神的情况下,对这些实施方式进行多种变化、修改、替换和变型,仍落入本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!