一种类蛇毒三肽的液相片段合成方法与流程
1.本发明属于多肽合成技术领域,具体涉及一种类蛇毒三肽的液相片段合成方法。
背景技术:
2.类蛇毒三肽(h-β-ala-pro-dab-nh-bzl)是一种模拟蛇毒毒素waglerin i活性的三肽,waglerin i发现于temple viper毒蛇的毒液,已被证明可抑制乙酰胆碱与乙酰胆碱受体的结合,从而抑制肌肉运动。类蛇毒肽是dsm荷兰皇家帝斯曼集团全资子公司pentapharm开发,其化学结构与毒蛇血清相似,具有抑制神经肌肉收缩,抚平肌肤皱纹的功能。应用于化妆品中,类蛇毒肽在减少动力性皱纹方面的功效是类肉毒杆菌的5倍,具有优秀的光滑肌肤、迅速祛皱的性能,分子结构如下所示:
[0003][0004]
目前,合成类蛇毒肽的方法主要有以下几种
[0005]
(1)液相逐个缩合:如专利cn 107936108 a,该方案以甲酯保护的氨基酸为原料,逐个缩合,最后进行c端修饰苄胺。该方法不断偶联、水解脱保护,步骤繁琐,偶联到后面工艺收率和纯度较低;此外,该方法用到了两次水解反应,pro-ome和dab-ome的水解,容易发生消旋;另外,所用原料均需要采用甲酯保护的氨基酸,成本较高。
[0006]
(2)固液相结合方式:该类方法一般先采用2-ctc resin固相合成全保护肽boc-β-ala-pro-dab(boc)-oh或fmoc-β-ala-pro-dab(boc)-oh,再在液相环境下与苄胺缩合(如专利cn 103570804 b)。该工艺粗肽纯度较高,但是使用2-ctc resin价格较高,而且氨基酸投料倍数基本都是大大过量,成本高,另外,固相合成在规模上也受限。
[0007]
(3)液相片段合成:如专利cn 107857797 a,该方法是合成片段h-pro-dab(boc)-nh-bzl,然后再与boc-β-ala-oh反应得到全保护肽。该片段合成策略简便易行,但使用的原料h-dab(boc)-oh价格较贵;另外,在fmoc-pro-dab(boc)-oh与苄胺偶联过程中,因dab(boc)主链氨基酸并非是氧羰基类保护基,在缩合反应中,容易产生dab的消旋,对产品质量有较大影响。
技术实现要素:
[0008]
针对现有技术的不足,本发明的目的在于提供一种类蛇毒三肽的液相片段合成方法。
[0009]
本发明提供一种类蛇毒三肽的液相片段合成方法,包括如下步骤:
[0010]
合成化合物z-β-ala-pro-oh;
[0011]
合成化合物fmoc-dab(z)-nhbzl;
[0012]
脱除化合物fmoc-dab(z)-nhbzl的fmoc保护基,得到化合物nh
2-dab(z)-nhbzl;
[0013]
以化合物z-β-ala-pro-oh和nh
2-dab(z)-nhbzl原料,合成化合物z-β-ala-pro-dab(z)-nhbzl;
[0014]
脱除化合物z-β-ala-pro-dab(z)-nhbzl的z保护基,得到化合物h-β-ala-pro-dab-nhbzl
·
2hbr;
[0015]
对h-β-ala-pro-dab-nhbzl
·
2hbr进行纯化转盐,得到h-β-ala-pro-dab-nhbzl。
[0016]
进一步地,所述化合物z-β-ala-pro-oh的合成方法为:以化合物z-β-ala-oh和honb或z-β-ala-oh和hosu为原料,在非质子性溶剂中,在缩合剂作用下进行活化反应,得到中间体z-β-ala-onb或z-β-ala-osu;在碱的作用下,中间体与h-pro-oh进行偶联反应,得到化合物z-β-ala-pro-oh。
[0017]
优选地,所述化合物z-β-ala-pro-oh的合成方法中:
[0018]
所述z-β-ala-oh和honb的摩尔比均为1:1.0~1.2;
[0019]
所述z-β-ala-oh和hosu的摩尔比为1:1.0~1.2;
[0020]
优选地,所述非质子性溶剂为n,n-二甲基甲酰胺、四氢呋喃、二恶烷、二甲基亚砜、乙腈、四氯化碳、甲苯、氯仿或二氯甲烷;
[0021]
优选地,所述非质子性溶剂为自n,n-二甲基甲酰胺;
[0022]
优选地,所述缩合剂为edc
·
hcl、dcc或dic;
[0023]
优选地,所述缩合剂为edc
·
hcl;
[0024]
优选地,所述活化反应的反应温度为室温,反应时间为2~4h;
[0025]
优选地,所述z-β-ala-onb或z-β-ala-osu和h-pro-oh的摩尔比为1:1.0~1.2;
[0026]
优选地,所述碱为碳酸氢钠、碳酸钠、碳酸氢钾、碳酸钾、三乙胺、n-甲基吗啡、dipea或2,4,6-三甲基吡啶;
[0027]
优选地,所述碱为碳酸氢钠;
[0028]
优选地,所述偶联反应的反应温度为室温,反应时间为2~4h。
[0029]
进一步地,所述化合物fmoc-dab(z)-nhbzl的合成方法为:以化合物fmoc-dab(z)-oh和苄胺为原料,在非质子性溶剂中,在偶联剂的作用下进行偶联反应,得到fmoc-dab(z)-nhbzl。
[0030]
优选地,所述化合物fmoc-dab(z)-nhbzl的合成方法中:
[0031]
所述fmoc-dab(z)-oh和苄胺的摩尔比为1:1.0~1.5;
[0032]
优选地,所述非质子性溶剂为n,n-二甲基甲酰胺、四氢呋喃、二恶烷、二甲基亚砜、乙腈、四氯化碳、甲苯、氯仿或二氯甲烷;
[0033]
优选地,所述非质子性溶剂为n,n-二甲基甲酰胺;
[0034]
优选地,所述偶联剂为化合物a、化合物a和dipea的组合物、化合物a和化合物b的组合物或化合物a和化合物b和dipea的组合物,所述化合物a为edc
·
hcl、dcc、dic、hatu、hbtu、tatu、tbtu、pyaop或pybop,所述化合物b为hoat或hobt;
[0035]
优选地,所述偶联剂为化合物a和dipea的组合物;
[0036]
优选地,所述化合物a和dipea的摩尔比为1:1.0~2.0;
[0037]
优选地,所述偶联剂为hbtu和和dipea的组合物;
[0038]
优选地,所述hbtu和dipea的摩尔比为1:1.0~2.0;
[0039]
优选地,所述偶联反应的反应温度为室温,反应时间为2~4h。
[0040]
进一步地,所述化合物fmoc-dab(z)-nhbzl的fmoc保护基在哌啶/二氯甲烷溶液或二乙胺/二氯甲烷溶液中脱除;
[0041]
优选地,所述哌啶/二氯甲烷溶液的体积分数为8%-20%;
[0042]
优选地,所述二乙胺/二氯甲烷溶液的体积分数为8%-20%;
[0043]
优选地,脱除fmoc保护基时,反应温度为室温,反应时间为1~2h。
[0044]
进一步地,所述化合物z-β-ala-pro-dab(z)-nhbzl的合成方法为:以化合物z-β-ala-pro-oh和nh
2-dab(z)-nhbzl为原料,在非质子性溶剂中,在偶联剂的作用下进行偶联反应,得到z-β-ala-pro-dab(z)-nhbzl。
[0045]
优选地,所述化合物z-β-ala-pro-dab(z)-nhbzl的合成方法中:
[0046]
所述z-β-ala-pro-oh和nh
2-dab(z)-nhbzl摩尔比为1:0.9~1.1;
[0047]
优选地,所述非质子性溶剂为n,n-二甲基甲酰胺、四氢呋喃、二恶烷、二甲基亚砜、乙腈、四氯化碳、甲苯、氯仿或二氯甲烷;
[0048]
优选地,所述非质子性溶剂为n,n-二甲基甲酰胺;
[0049]
优选地,所述偶联剂为化合物a和化合物b的组合物或化合物a和化合物b和dipea的组合物,其中,所述化合物a为edc
·
hcl、dcc、dic、hatu、hbtu、tatu、tbtu、pyaop或pybop,所述化合物b为hoat或hobt;
[0050]
优选地,所述偶联剂为化合物a和化合物b和dipea的组合物;
[0051]
优选地,所述偶联剂为化合物a和化合物b和dipea的组合物;
[0052]
优选地,所述化合物a和化合物b和dipea的摩尔比为1:1:1.0~2.0
[0053]
优选地,所述偶联剂为edc
·
hcl、hoat和dipea的组合物;
[0054]
优选地,所述edc
·
hcl、hoat和dipea的摩尔比为=1:1:1.0~2.0;
[0055]
优选地,所述偶联反应的反应温度为室温,反应时间为3~5h。
[0056]
进一步地,所述化合物h-β-ala-pro-dab-nhbzl
·
2hbr的z保护基在溴化氢/醋酸溶液中脱除;
[0057]
优选地,所述溴化氢/醋酸溶液的体积分数为33%;
[0058]
优选地,脱除z保护基时,反应温度为室温,反应时间为3~5h。
[0059]
进一步地,所述h-β-ala-pro-dab-nhbzl
·
2hbr进行纯化转盐的具体步骤如下:
[0060]
将得到的h-β-ala-pro-dab-nhbzl
·
2hbr粗品用水溶解并过滤,采用c18反相柱,以0.3%tfa为a相,纯乙腈为b相进行梯度洗脱,得到流动相,再以0.5%醋酸为a相,纯乙腈为b相进行转盐,收集洗脱液并冻干,即得h-β-ala-pro-dab-nhbzl。
[0061]
本发明的有益效果为:
[0062]
(1)本发明提供的类蛇毒三肽的液相片段合成方法采用2+2片段合成策略,先合成化合物z-β-ala-pro-oh和nh
2-dab(z)-nhbzl,然后以化合物z-β-ala-pro-oh和nh
2-dab(z)-nhbzl原料进行h-β-ala-pro-dab-nhbzl的合成,该工艺路线短,操作简便快捷,并且两片段能够通过析晶分离,提高了合成的效率,利于工业化生产。
[0063]
(2)本发明以z-β-ala-oh,fmoc-dab(z)-oh为原料,避免使用强碱如氢氧化钠或氢氧化锂来处理氨基酸酯的反应,减少反应步骤,降低消旋杂质产生,提高了中间体纯度。
[0064]
(3)该合成方法所用原料价格便宜,合成成本较低,比如使用的fmoc-dab(z)-oh,
其价格比常用来合成类蛇毒肽的h-dab(boc)-oh、h-dab(boc)-ome和fmoc-dab(boc)-oh等便宜。
附图说明
[0065]
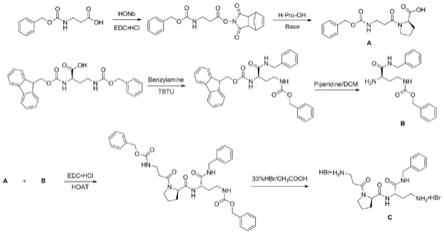
图1为一个具体实施方案的类蛇毒三肽的液相片段的合成流程;
[0066]
图2为实施例1中z-β-ala-pro-oh的质谱;
[0067]
图3为实施例2中fmoc-dab(z)-nhbzl的质谱;
[0068]
图4为实施例2中nh
2-dab(z)-nhbzl的质谱;
[0069]
图5为实施例3中z-β-ala-pro-dab(z)-nhbzl的质谱;
[0070]
图6为实施例3中h-β-ala-pro-dab-nhbzl
·
2hbr的质谱;
[0071]
图7为实施例3中h-β-ala-pro-dab-nhbzl的检测谱。
具体实施方式
[0072]
为了更清楚地理解本发明,现参照下列实施例及附图进一步描述本发明。实施例仅用于解释而不以任何方式限制本发明。实施例中,各原始试剂材料均可商购获得,未注明具体条件的实验方法为所属领域熟知的常规方法和常规条件,或按照仪器制造商所建议的条件。
[0073]
本发明缩写及英文含义如下表。
[0074]
缩写及英文含义z苄氧羰基fmoc9-芴基甲氧基羰基boc叔丁氧羰基honbn-羟基-5-降冰片烯-2,3-二甲酰亚胺edc
·
hcl1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐tbtu2-(1h-苯并三偶氮l-1-基)-1,1,3,3-四甲基脲四氟硼酸酯dipean,n-二异丙基乙胺hoatn-羟基-7-氮杂苯并三氮hbtuo-(苯并三唑-1-基)-n,n,n',n'-四甲基脲六氟磷酸酯hosun-羟基丁二酰亚胺dcc二环己基碳二亚胺dicn,n'-二异丙基碳二亚胺hatuo-(7-氮杂苯并三唑-1-基)-n,n,n',n'-四甲基脲六氟磷酸盐tatu2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲四氟硼酸盐tbtu2-(1h-苯并三偶氮l-1-基)-1,1,3,3-四甲基脲四氟硼酸酯pyaop六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷pybop1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐hobt1-羟基苯并三唑
[0075]
图1显示了一个具体实施方案的类蛇毒三肽的液相片段的合成流程。
[0076]
实施例1:合成z-β-ala-pro-oh
dab-nhbzl,总收率为67.0%,纯度为95.48%(见图7)。
[0091]
实施例4:合成z-β-ala-pro-oh
[0092]
与实施例1不同之处在于,所述的honb替换为hosu,获得14.45g z-β-ala-osu,收率为90.3%,纯度为95.7%,得到12.18g z-β-ala-pro-oh,收率为84.3%,纯度为94.6%。
[0093]
实施例5:合成并制备h-β-ala-pro-dab-nhbzl
[0094]
与实施例3不同之处在于,所述z-β-ala-pro-oh的原料取自实施例4,制备后的获得9.77g h-β-ala-pro-dab-nhbzl,总收率为64.6%,纯度为95.1%。
[0095]
实施例6:合成nh
2-dab(z)-nhbzl
[0096]
与实施例2(2)不同之处在于,所述的哌啶/二氯甲烷溶液替换为二乙胺/二氯甲烷溶液,合成并制备得到13.52gnh
2-dab(z)-nhbzl,收率为87.5%,纯度为91.37%。
[0097]
实施例7:合成并制备h-β-ala-pro-dab-nhbzl
[0098]
与实施例3不同之处在于,其nh
2-dab(z)-nhbzl原料取自实施例6,制备后获得9.07gh-β-ala-pro-dab-nhbzl,总收率为59.9%,纯度为96.2%。
[0099]
实施例8:合成z-β-ala-pro-oh
[0100]
与实施例1不同之处在于,饱和碳酸氢钠水溶液替换为碳酸钠(与h-pro-oh同当量)水溶液,制备后得到13.98g z-β-ala-pro-oh,收率为90.7%,纯度为91.07%。
[0101]
实施例9:合成并制备h-β-ala-pro-dab-nhbzl
[0102]
与实施例3不同之处在于所述z-β-ala-pro-oh原料选自实施例8,制备后获得8.68gh-β-ala-pro-dab-nhbzl,总收率为57.4%,纯度为95.0%。
[0103]
实施例10:合成z-β-ala-pro-oh
[0104]
与实施例1不同之处在于,饱和碳酸氢钠水溶液替换为dipea(z-β-ala-onb的2倍当量)水溶液,制备后得到13.12g z-β-ala-pro-oh,收率为85.1%,纯度为92.15%。
[0105]
实施例11:合成并制备h-β-ala-pro-dab-nhbzl
[0106]
与实施例3不同之处在于所述z-β-ala-pro-oh原料选自实施例10,制备后获得8.28gh-β-ala-pro-dab-nhbzl,总收率为54.8%,纯度为95.2%。
[0107]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1