一种西地那非中间体的制备方法与流程
1.本发明属于药物合成技术领域,具体涉及一种西地那非中间体的制备方法。
背景技术:
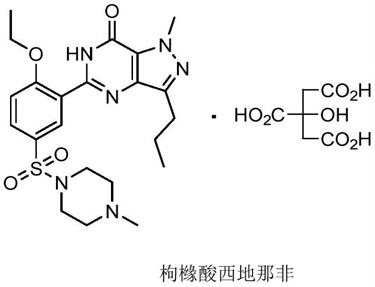
2.西地那非是一种5型磷酸二酯酶(pde-5)的选择性抑制剂,而pde-5是对cgmp专属的一种磷酸二酯酶亚型,是高效治疗男性勃起功能障碍(ed)的药物。西地那非的化学名为:1-甲基-3-正丙基-5-[2-乙氧基-5-(4-甲基哌嗪-1-磺酰基)苯基]-1,6-二氢-7h-吡唑并[4,2-d]吡啶-7-酮枸橼酸盐,一般以其商业名称viagra广为人知。枸橼酸西地那非的化学结构如下所示:
[0003][0004]
西地那非主要的合成路线以4-氨基-1-甲基-3-正丙基吡唑-5-甲酰胺(式i)和2-乙氧基苯甲酰氯或2-乙氧基苯甲酰酸为起始原料,经缩合反应、嘧啶酮环合反应、氯磺酰化再与n-甲基哌嗪缩合,成盐得到枸橼酸西地那非。
[0005]
[0006]
因此,4-氨基-1-甲基-3-正丙基吡唑-5-甲酰胺(式:i),是合成西地那非的一个关键中间体。
[0007][0008]
文献报道的该中间体的合成路线是以2-戊酮为原料,在乙醇钠存在下与草酸二乙酯缩后,与水合肼环合得到吡唑环,再与硫酸二甲酯反应进行甲基化,进行水解得到羧酸,后用混酸硝化得到硝基羧酸物,再使用氯化亚砜氯化,得到酰氯后氨解得酰胺,最后还原硝基得到4-氨基-1-甲基-3-正丙基吡唑-5-甲酰胺。反应式如下所示:
[0009][0010]
该合成路线涉及7个中间体,其以2-戊酮与草酸二乙酯反应得到中间体1、经过水合肼环合得到中间体2、中间体2经过使用硫酸二甲酯进行n-甲基化得到中间体3、中间体3再经过水解、硝化、酰氯化、酰胺化、还原得到中间体7(式:i)。
[0011]
上述现有技术中,中间体3的制备使用硫酸二甲酯进行n-甲基化,硫酸二甲酯属高毒物,在工业生产过程中大量使用硫酸二甲酯可能会造成严重的污染和中毒事件。为了避免在生产过程中使用硫酸二甲酯可能会造成严重的污染和中毒事件的发生,本发明进行了制备方法的技术改造。
技术实现要素:
[0012]
为了解决上述技术问题,本发明提供如下技术方案。
[0013]
一种合成西地那非的中间体式i的制备方法,包括如下步骤(1):在4-二甲氨基吡啶与四丁基溴化铵的存在下,3-正丙基-1h-吡唑-5-羧酸乙酯与磷酸三甲酯进行n-甲基化反应,得到中间体-1即1-甲基-3-正丙基-1h-吡唑-5-羧酸乙酯,
[0014]
根据本发明的制备方法,上述反应中,其反应温度为85~110℃,优选95~100℃。
[0015]
根据本发明的制备方法,其反应压力为表压0.10~0.20mpa。优选的,反应压力为表压0.10~0.15mpa。
[0016]
根据本发明的制备方法,3-正丙基-1h-吡唑-5-羧酸乙酯与4-二甲基吡啶的摩尔用量比0.8~1.2∶1,磷酸三甲酯的摩尔用量为3-正丙基-1h-吡唑-5-羧酸乙酯的1.5-3倍,四丁基溴化铵的用量为3-正丙基-1h-吡唑-5-羧酸乙酯的0.1~0.3倍。优选的,3-正丙基-1h-吡唑-5-羧酸乙酯∶磷酸三甲酯∶四丁基溴化铵∶4-二甲基吡啶的摩尔用量比为1∶2∶0.1∶1.1。
[0017]
根据本发明的制备方法,所述n-甲基化反应完成后,以碱液中和,二氯甲烷提取,浓缩,减压蒸馏得到中间体-1。
[0018]
根据本发明的制备方法,包括如下步骤,以得到中间体式i化合物:
[0020][0021]
有益效果:
[0022]
本发明方法改进了制备1-甲基-3-正丙基-1h-吡唑-5-羧酸乙酯(中间体-1)的反应试剂。现有技术中使用硫酸二甲酯与3-正丙基-1h-吡唑-5-羧酸乙酯进行n-甲基化反应,硫酸二甲酯属于高毒性性类试剂。本发明采用了低毒性的甲基化试剂磷酸三甲酯,并且研究了反应条件,发现在助剂存在下,调整反应压力,使用低毒性磷酸三甲酯可制得产率很高的中间体-1。
具体实施方式
[0023]
下文将结合具体实施例对本发明的制备方法做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。除非另有说明,
以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
[0024]
1-甲基-3-正丙基-1h-吡唑-5-羧酸乙酯(中间体-1)的制备:
[0025]
向5l压力釜中加入3-正丙基-1h-吡唑-5-羧酸乙酯(550g,3.00mol)、新蒸馏的磷酸三甲酯(841g,6.00mol),室温搅拌至溶解完全,加入4-二甲氨基吡啶110g(0.90mol)与四丁基溴化铵97g(0.30mol),拧紧反应釜盖。搅拌下慢慢加热,稳定内温至95~100℃后,用排气阀与氮气调节表压约0.10~0.15mpa,保温搅拌反应15小时,冷却,慢慢泄掉压力。
[0026]
将反应液转移至5l反应瓶中,冷却下用饱和碳酸钠溶液中和反应,当无气泡溢出后,使用二氯甲烷500ml
×
3萃取,合并二氯甲烷萃取液,在40~60℃常压蒸馏出二氯甲烷,减压蒸馏掉过量的磷酸三甲酯60~90℃/5~8mmhg,收集主馏分150~170℃/5~8mmhg,得到中间体-1无色油状物488.7g,收率:83.0%。
[0027]
本发明在该实施例的操作步骤下,进行了一系列反应条件的研究,实验条件和实验结果如下表所示:
[0028]
表1
[0029][0030]
表2
[0031][0032]
表3
[0033][0034][0035]
中间体-2的制备:
[0036]
向5l三口瓶中加入中间体-1(392g,2.00mol),加入6n氢氧化钠水溶液970ml,加热至回流反应1.5小时,降温至40℃以下,加入水970ml,滴加6n盐酸970ml进行中和,滴加完毕后,析出固体,搅拌下降温至15~20℃,保温搅拌1小时,抽滤,500
×
3ml纯化水洗涤,抽干,固体在50~55℃鼓风干燥12小时,得白色固体293g,收率:87.1%。熔点:151~153℃。
[0037]
中间体-3的制备:
[0038]
向3l三口瓶中加入浓硫酸450ml与发烟硝酸382ml,搅拌下控制内温65℃以下,分5批次加入中间体-2(280g,1.66mol),加完后在60~65℃保温反应15小时,冷却至室温,将反应液倒入装有碎冰1200g的塑料桶中,边倒边搅拌,析出白色固体,冷却至室温,抽滤,固体用冷水300ml
×
3次洗涤后,抽干,湿物料在50~55℃鼓风干燥15小时,得白色固体346g,收率:97.5%。熔点:122~124℃。
[0039]
中间体-4的制备:
[0040]
向3l三口瓶中加入中间体-3(330g,1.55mol),加入二氯亚砜990ml,慢慢加热回流,搅拌反应4小时。降温至40~55℃,减压浓缩掉过量的二氯亚砜,加入丙酮660ml,搅拌溶解剩余物。
[0041]
将溶解物慢慢倒入装有碎冰990g与浓氨水990ml的塑料桶中,边倒边搅拌,析出淡黄色固体(ph≈10),搅拌10分钟,抽滤,固体用冷水250ml次洗涤后,抽干,湿物料在50~55℃鼓风干燥15小时,得淡黄色固体280g,收率:75.5%。熔点:142~143℃。
[0042]
中间体-5的制备:
[0043]
向3l三口瓶中加入中间体-4(260g,1.23mol)、乙醇1050ml,搅拌下加入二水氯化
亚锡(833g,3.69mol),加热至回流反应4小时,撤掉热浴,降温至30℃以下,冰水浴控制内温30℃以下,用2n氢氧化钠调节至ph≈10,冷却至室温,使用二氯甲烷1050ml
×
3提取,合并提取液,滤液用500ml纯化水洗涤一次,分取二氯甲烷相,减压浓缩,室温下向剩余物中加入异丙醚520ml,搅拌1小时,过滤析出固体,使用冷的异丙醚200ml洗涤滤饼一次,抽干,湿物料在40~50℃鼓风干燥12小时,得到类白色固体196g,收率:87.5%。熔点:99~101℃。
[0044]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1