双面粘合带的制作方法
本发明涉及一种虽然为薄型但是冲击吸收性和再剥离性优异的双面粘合带。
背景技术:
就移动电话、便携式信息终端(personaldigitalassistants、pda)等便携电子设备而言,对部件的固定配置或设备本体的设计进行了研究,以便即使考虑到从使用者的手边掉落至脚下而施加冲击也不会使部件脱落或破损。因此,作为用于将部件固定于设备本体而使用的双面粘合带,也期望即使在施加冲击的情况下部件也不会脱落、且不会对部件施加强冲击的双面粘合带。
作为将构成便携电子设备的部件固定于设备本体的冲击吸收带,研究了例如具有由聚烯烃发泡体构成的基材的双面粘合带。
在专利文献1和2中记载了一种冲击吸收带,其在基材层的至少单面层叠丙烯酸系粘合剂层而一体化,基材层为具有特定的交联度和气泡的纵横比的交联聚烯烃系树脂发泡片。
另外,在将汽车构件(例如车载用面板)固定于汽车本体的用途中也使用双面粘合带,作为这样的双面粘合带,也使用冲击吸收性能优异的具有由聚烯烃发泡体构成的基材的双面粘合带。
近年来,便携电子设备部件或车载用电子设备部件的进一步薄型化不断推进,所使用的双面粘合带也要求薄型化。然而,若将具有由以往的发泡体构成的基材的双面粘合带薄化,则存在基材的强度降低而容易引起条带的层间剥离的问题。另外,若基材的强度降低,则在将条带拉伸时导致条带过度地伸展,因此还存在再剥离性差的问题。
现有技术文献
专利文献
专利文献1:日本特开2009-242541号公报
专利文献2:日本特开2009-258274号公报
技术实现要素:
发明要解决的问题
本发明的目的在于提供虽然为薄型但是冲击吸收性和再剥离性优异的双面粘合带。
用于解决问题的手段
本发明为一种双面粘合带,其是在由聚烯烃发泡体构成的基材的双面具有丙烯酸类粘合剂层的双面粘合带,上述由聚烯烃发泡体构成的基材的厚度为50~120μm,上述由聚烯烃发泡体构成的基材在md方向及td方向的平均气泡直径为30μm以上且70μm以下,双面粘合带的总厚度为80~150μm。
以下对本发明进行详细叙述。
本发明人等对因薄型化导致的基材强度降低的问题进行了深入研究,结果发现:在由聚烯烃发泡体构成的基材的双面具有丙烯酸类粘合剂层的双面粘合带中,通过使基材的平均气泡直径小于以往的基材,从而即使在减薄基材的情况下,也能确保强度。其结果发现:虽然具有由发泡体构成的基材,但仍得到薄型、且冲击吸收性和再剥离性优异的双面粘合带,以至完成本发明。
本发明的双面粘合带具有由聚烯烃发泡体构成的基材(以下也简称为“基材”。)。通过使用聚烯烃发泡体作为基材,可以对所得的双面粘合带赋予冲击吸收性。
上述聚烯烃发泡体只要是满足后述的md方向及td方向的平均气泡直径的聚烯烃发泡体则并无特别限定,可列举例如聚乙烯系发泡体、聚丙烯系发泡体、乙烯-丙烯系发泡体等。其中,优选为聚乙烯系发泡体。
上述基材的厚度的下限为50μm、上限为120μm。本发明的双面粘合带虽然具有由发泡体构成的基材,但即使在厚度薄的情况下也能确保基材的强度,因此即使薄型化也不易引起层间剥离,再剥离性也优异。上述基材的厚度的优选的下限为60μm、优选的上限为100μm。
上述聚烯烃发泡体在md方向及td方向的平均气泡直径为30μm以上且70μm以下。
通过使上述聚烯烃发泡体具有30μm以上且70μm以下这样的小的平均气泡直径,从而即使在减薄基材的情况下也能保持强度,能够制成兼顾冲击吸收性和再剥离性、且薄型的双面粘合带。上述聚烯烃发泡体的平均气泡直径的优选的下限为35μm,更优选的下限为40μm。另外,上述平均气泡直径的优选的上限为65μm,更优选的上限为55μm。
作为减小上述平均气泡直径的方法,可列举在发泡前对树脂进行加压的方法等。通过在发泡前对树脂进行加压,可以在发泡时抑制气泡的生长。另外,发泡时向md方向及td方向的拉伸率越小,越能减小md方向及td方向的气泡直径,因此通过在发泡前进行加压来减薄树脂的厚度,从而能够制成既抑制发泡时的拉伸又薄的基材,并且能够减小气泡。
需要说明的是,md方向(machinedirection)是指将聚烯烃发泡体挤出加工为片状时的挤出方向,td方向(transversedirection)是指与md方向垂直的方向。
上述md方向的平均气泡直径可以利用以下的方法来测定。
首先,将聚烯烃发泡体的样品切割成50mm见方,在液氮中浸渍1分钟后,用剃须刀片沿着与md方向及厚度方向平行的面进行切割。接着,使用数字显微镜(例如keyence公司制、“vhx-900”等),以200倍倍率拍摄放大照片,对在md方向的长度2mm的切割面上存在的全部气泡测定md方向的气泡直径。将该操作反复5次,将全部的md方向的气泡直径的平均值设为md方向的平均气泡直径。
除了将聚烯烃发泡体的样品沿着与td方向及厚度方向平行的面进行切割以外,上述td方向的平均气泡直径也可以同样地进行测定。
需要说明的是,在md方向及td方向不明确的情况下,也可以如以下所示地确定上述md方向的平均气泡直径及上述td方向的平均气泡直径。
即,首先,在聚烯烃发泡体的样品中,选择与度方向垂直的任意一个方向作为第一方向,将与第一方向及厚度方向垂直的方向作为第二方向。接着,将聚烯烃发泡体的样品沿着与上述第一方向及厚度方向平行的面进行切割,与上述的测定方法同样地测定上述第一方向的平均气泡直径。将所得的平均气泡直径作为md方向的平均气泡直径来对待。进而,除了将聚烯烃发泡体的样品沿着与上述第二方向及厚度方向平行的面进行切割以外,与上述的测定方法同样地测定,将所得的平均气泡直径作为td方向的平均气泡直径来对待。
优选在上述聚烯烃发泡体的厚度方向包含1个或2个上述聚烯烃发泡体中的气泡。通过在上述基材的厚度方向包含1个或2个具有上述平均气泡直径的气泡,从而即使在减薄基材的情况下也容易确保强度。
优选上述聚烯烃发泡体的发泡倍率的下限为1.4cm3/g、上限为2.0cm3/g。通过使上述基材的发泡倍率为1.4cm3/g以上,可以提高双面粘合带的柔软性和冲击吸收性。通过使上述基材的发泡倍率为2.0cm3/g以下,可以得到基材的强度提高、能够防止层间剥离且再剥离性优异的双面粘合带。
需要说明的是,发泡倍率可以按照依据jisk-6767而使用电子比重计(例如mirage公司制、“ed120t”)测定得到的密度的倒数来计算。
上述md方向及td方向的平均气泡直径为30μm以上且70μm以下的、由聚烯烃发泡体构成的基材例如可以通过经过如以下那样的工序来制造。
(1)将聚烯烃系树脂、热分解型发泡剂及其他添加剂供给至挤出机,进行熔融混炼,并从挤出机挤出为片状,由此得到成为片状的聚烯烃系树脂组合物的工序。
(2)将成为片状的聚烯烃系树脂组合物进行交联的工序。
(3)对交联的片状的聚烯烃系树脂组合物进行加压,之后进行加热,使热分解型发泡剂发泡,并向md方向或td方向中的任意一方或双方进行拉伸的工序。
需要说明的是,作为聚烯烃发泡体的制造方法,除该方法外,还可利用国际公开第2005/007731号记载的方法来制造。
作为上述聚烯烃系树脂,可列举例如聚乙烯系树脂、聚丙烯系树脂等,其中,优选为聚乙烯系树脂。
上述聚乙烯系树脂可以为乙烯均聚物,但优选为通过将乙烯与少量的α-烯烃共聚而得到的乙烯-α-烯烃共聚物。通过使上述聚乙烯系树脂为乙烯与少量的α-烯烃的共聚物,可以提高发泡体的柔软性并进一步提高冲击吸收性。在上述乙烯-α-烯烃共聚物中,更优选直链状低密度聚乙烯。
作为上述乙烯-α-烯烃共聚物中的α-烯烃,可列举例如丙烯、1-丁烯、1-戊烯、4-甲基-1-戊烯、1-己烯、1-庚烯及1-辛烯等。其中,优选碳原子数4~10的α-烯烃。
上述乙烯-α-烯烃共聚物中的α-烯烃的优选的上限为30重量%,更优选的上限为10重量%。
作为上述聚乙烯系树脂,也优选乙烯-乙酸乙烯酯共聚物。乙烯-乙酸乙烯酯共聚物通常为含有50重量%以上的来源于乙烯的结构单元的共聚物。
从提高发泡体的柔软性、提高冲击吸收性的观点出发,上述聚乙烯系树脂优选为低密度。上述聚乙烯系树脂的密度优选为0.920g/cm3以下,更优选为0.880~0.915g/cm3,进一步优选为0.885~0.910g/cm3。
需要说明的是,密度为依据astmd792测定得到的值。
作为成为上述聚丙烯系发泡体的原料的聚丙烯系树脂,可列举例如丙烯均聚物、含有50重量%以上的来源于丙烯的结构单元的丙烯-α-烯烃共聚物等。它们可以单独使用,也可以并用2种以上。
作为上述丙烯-α-烯烃共聚物中的α-烯烃,可列举例如乙烯、1-丁烯、1-戊烯、4-甲基-1-戊烯、1-己烯、1-庚烯、1-辛烯等。其中,优选为碳数6~12的α-烯烃。
从提高柔软性和冲击吸收性的观点出发,上述聚烯烃系树脂组合物中的聚烯烃系树脂优选含有通过使用茂金属化合物、齐格勒-纳塔化合物、氧化铬化合物等作为催化剂而聚合得到的聚乙烯系树脂、聚丙烯系树脂或它们的混合物,更优选为直链状低密度聚乙烯。
上述茂金属化合物优选为具有以π电子系的不饱和化合物夹持过渡金属的结构的双(环戊二烯)金属络合物等化合物。具体而言,可列举例如对于钛、锆、镍、钯、铪及铂等四价过渡金属存在1个或2个以上的环戊二烯环或其类似物作为配体(配位体)的化合物。
此种茂金属化合物的活性点的性质均匀,各活性点具备相同的活性度。其结果为:就使用上述茂金属化合物而合成的聚合物而言,由于分子量、分子量分布、组成、组成分布等的均匀性高,因此在将包含使用上述茂金属化合物而合成的聚合物的片材交联的情况下,交联均匀地进行。经均匀地交联的片材容易均匀地拉伸,因此容易使聚烯烃发泡体的厚度变得均匀。
作为配体,可列举例如环戊二烯环、茚环等环式化合物。上述环式化合物可以具有烃基、取代烃基或烃-取代类金属基等取代基。作为上述烃基,可列举例如甲基、乙基、各种丙基、各种丁基、各种戊基、各种己基、2-乙基己基、各种庚基、各种辛基、各种壬基、各种癸基、各种鲸蜡基、苯基等。需要说明的是,在此,“各种”是指正-、仲-、叔-、异-等各种异构体。
另外,也可以使用将上述环式化合物作为低聚物聚合而得的物质作为配体。
进而,除了π电子系的不饱和化合物以外,还可以使用氯或溴等一价阴离子配体、二价阴离子螯合物配体、烃、醇盐、芳基酰胺、芳基醚、酰胺、芳基酰胺、磷化物、芳基磷化物等。
作为上述包含四价过渡金属、配体的茂金属化合物,可列举例如环戊二烯钛三(二甲基酰胺)、甲基环戊二烯钛三(二甲基酰胺)、双(环戊二烯)二氯化钛、二甲基甲硅烷基四甲基环戊二烯叔丁基酰胺二氯化锆等。
上述茂金属化合物通过与特定的共催化剂(助催化剂)组合而在各种烯烃的聚合时发挥作为催化剂的作用。作为上述共催化剂,可列举甲基铝氧烷(mao)、硼系化合物等。上述共催化剂相对于上述茂金属化合物的使用比例优选为10~100万摩尔倍,更优选为50~5,000摩尔倍。
在使用通过将上述茂金属化合物用作催化剂而得的聚乙烯系树脂、乙烯-乙酸乙烯酯共聚物或它们的混合物的情况下,其含量优选为聚烯烃系树脂全体的40重量%以上,更优选为50重量%以上,进一步优选为60重量%以上,特别优选为100重量%。通过使将上述茂金属化合物用作催化剂而得的聚乙烯系树脂、乙烯-乙酸乙烯酯共聚物或它们的混合物的含量为40重量%以上,从而即使在上述聚烯烃发泡体的厚度薄的情况下,也能得到高压缩强度。
上述齐格勒-纳塔化合物优选使用作为三乙基铝-四氯化钛固体复合物、利用以下方法制造的齐格勒-纳塔化合物,所述方法为:使将四氯化钛用有机铝化合物还原、再用各种电子供体及电子受体进行处理而得的三氯化钛组合物、有机铝化合物和芳香族羧酸酯组合的方法(参照日本特开昭56-100806号、日本特开昭56-120712号、日本特开昭58-104907号的各公报);使四氯化钛和各种电子供体与卤化镁接触的担载型催化剂的方法(参照日本特开昭57-63310号、日本特开昭63-43915号、日本特开昭63-83116号的各公报)等。
上述聚烯烃系树脂组合物可以包含除上述的聚烯烃系树脂以外的树脂等任意成分。
作为上述任意成分,可列举除聚烯烃系树脂以外的树脂、橡胶。这些任意成分优选为合计量少于聚烯烃系树脂的含量,具体而言,相对于聚烯烃系树脂100重量份,优选为50重量份以下,更优选为30重量份以下。
上述热分解型发泡剂并无特别限制,可列举例如偶氮二甲酰胺、n,n’-二亚硝基五亚甲基四胺、对甲苯磺酰基氨基脲等,其中,优选偶氮二甲酰胺。上述热分解型发泡剂可以单独使用,也可以组合使用2种以上。
上述聚烯烃系树脂组合物中的上述热分解型发泡剂的含量相对于聚烯烃系树脂100重量份优选为1~12重量份,更优选为1~8重量份。通过使上述热分解型发泡剂的含量为上述范围内,从而上述聚烯烃系树脂组合物的发泡性提高,容易得到具有所期望的发泡倍率的聚烯烃发泡体,并且可以提高拉伸强度和压缩恢复性。
作为上述添加剂,可列举分解温度调节剂、交联助剂、抗氧化剂等。
上述分解温度调节剂是通过降低热分解型发泡剂的分解温度或加快分解速度而调整发泡体的表面状态等的制剂。
作为分解温度调节剂,可列举例如氧化锌、硬脂酸锌、尿素等。
上述分解温度调节剂相对于上述聚烯烃系树脂100重量份的含量优选为0.01~5重量份。
上述交联助剂是通过在聚烯烃系树脂中进行添加而在将后述的聚烯烃系树脂组合物进行交联的工序中降低所照射的电离射线量、防止与电离射线的照射相伴的树脂分子的切割、劣化的制剂。
作为上述交联助剂,可列举例如多官能单体等。具体而言,可列举:三羟甲基丙烷三甲基丙烯酸酯、三羟甲基丙烷三丙烯酸酯、偏苯三酸三烯丙基酯、1,2,4-苯三甲酸三烯丙基酯、异氰脲酸三烯丙基酯等在1分子中具有3个官能团的化合物;1,6-己二醇二甲基丙烯酸酯、1,9-壬二醇二甲基丙烯酸酯、1,10-癸二醇二甲基丙烯酸酯、二乙烯基苯等在1分子中具有2个官能团的化合物;邻苯二甲酸二烯丙基酯、对苯二甲酸二烯丙基酯、间苯二甲酸二烯丙基酯、乙基乙烯基苯、新戊二醇二甲基丙烯酸酯、甲基丙烯酸月桂基酯、甲基丙烯酸硬脂基酯等。
这些交联助剂可以单独使用,也可以组合使用2种以上。
交联助剂的添加量相对于聚烯烃系树脂100重量份优选为0.2~10重量份,更优选为0.3~5重量份,进一步优选为0.5~5重量份。通过使上述交联助剂的添加量为0.2重量份以上,可以稳定地得到具有所期望的交联度的聚烯烃发泡体。通过使上述交联助剂的添加量为10重量份以下,可以容易地控制聚烯烃发泡体的交联度。
为了防止由热所致的氧化劣化而配合上述抗氧化剂。作为上述抗氧化剂,可列举2,6-二-叔丁基对甲酚等酚系抗氧化剂等。
在将上述聚烯烃系树脂组合物进行交联的工序中,作为将聚烯烃系树脂组合物进行交联的方法,可列举例如:对聚烯烃系树脂组合物照射电子射线、α射线、β射线、γ射线等电离射线的方法;在形成聚烯烃系树脂组合物时预先配合有机过氧化物,之后,对聚烯烃系树脂组合物进行加热而使有机过氧化物分解的方法。这些方法可以单独使用,也可以并用2种以上,但是,从均质地进行交联的观点出发,优选照射电离射线的方法。
上述照射电离射线的方法中的电离射线的照射量优选以使凝胶分数达到5~45重量%的方式进行调节。作为具体的照射量,优选0.5~20mrad,更优选3~12mrad。
需要说明的是,聚烯烃系树脂组合物的凝胶分数(交联度)可以按照以下方式来测定。
即,从聚烯烃发泡体采集约50mg的试验片,并对试验片的重量a(mg)进行精确称量。接着,将该试验片浸渍于105℃的二甲苯30cm3中,放置24小时后,用200目的金属丝网进行过滤,采集金属丝网上的不溶解成分,进行真空干燥,并对不溶解成分的重量b(mg)进行精确称量。根据下述式由所得的值计算凝胶分数(重量%)。
凝胶分数(重量%)=(b/a)×100
作为将有机过氧化物预先配合于上述聚烯烃系树脂组合物的方法中的有机过氧化物,可列举例如1,1-双(叔丁基过氧化)3,3,5-三甲基环己烷、1,1-双(叔丁基过氧化)环己烷等。它们可以单独使用1种,也可以并用2种以上。
上述有机过氧化物的添加量相对于聚烯烃系树脂100重量份优选为0.01~5重量份,更优选为0.1~3重量份。通过使上述有机过氧化物的添加量为上述范围内,从而容易进行聚烯烃系树脂组合物的交联,并且可以抑制在所得的聚烯烃发泡体中存在的有机过氧化物的分解残渣的量。
在使上述聚烯烃系树脂组合物发泡的工序中,聚烯烃系树脂组合物的发泡方法并无特别限定,可列举例如对聚烯烃系树脂组合物利用热风进行加热的方法、利用红外线进行加热的方法、利用盐浴进行加热的方法、利用油浴进行加热的方法等,这些方法也可以并用。
需要说明的是,聚烯烃系树脂组合物的发泡并不限定于使用热分解型发泡剂的例子,也可以使用基于丁烷气体等的物理发泡。
在将上述聚烯烃系树脂组合物进行拉伸的工序中,拉伸可以在使聚烯烃系树脂组合物发泡而得到发泡体后进行,也可以在使聚烯烃系树脂组合物发泡的同时进行。需要说明的是,在使聚烯烃系树脂组合物发泡而得到发泡体后对发泡体进行拉伸的情况下,优选的是在不冷却发泡体而维持发泡时的熔融状态的状态下持续地拉伸发泡体,也可以在将冷却后的发泡体再度加热而成为熔融或软化状态后对发泡体进行拉伸。
上述聚烯烃系树脂组合物在md方向的拉伸倍率优选为1.1~3.0倍,更优选为1.7~2.8倍。通过使聚烯烃发泡体在md方向的拉伸倍率为上述下限值以上,从而容易使聚烯烃发泡体的柔软性和拉伸强度变得良好。另外,通过使拉伸倍率为上限值以下,从而防止发泡体在拉伸中发生断裂的情况、或从发泡中的发泡体脱除发泡气体而使发泡倍率降低的情况,使聚烯烃发泡体的柔软性和拉伸强度变得良好,容易成为品质也均匀的聚烯烃发泡体。另外,聚烯烃发泡体也可以在td方向也以上述范围的拉伸倍率进行拉伸。
本发明的双面粘合带在基材的双面具有丙烯酸类粘合剂层。
关于上述丙烯酸类粘合剂层的厚度,优选单面的丙烯酸类粘合剂层的厚度为50μm以下。通过使上述丙烯酸类粘合剂层的厚度为50μm以下,可以兼顾双面粘合带的耐冲击性及剪切粘合力与双面粘合带的薄度。上述丙烯酸类粘合剂层的更优选的上限为30μm。上述丙烯酸类粘合剂层的厚度的下限并无特别限制,但优选为8μm,更优选为10μm。
构成上述丙烯酸类粘合剂层的丙烯酸类共聚物优选通过将包含丙烯酸丁酯和丙烯酸2-乙基己酯的单体混合物共聚而得到。
在全部单体混合物中所占的丙烯酸丁酯的优选含量为40~80重量%。通过使丙烯酸丁酯的含量为40重量%以上,从而使上述丙烯酸类粘合剂层处于适度的柔软度和内聚力,可以提高双面粘合带的剪切粘合力。通过使丙烯酸丁酯的含量为80重量%以下,从而使上述丙烯酸类粘合剂层处于适度的硬度,粘合力或粘性提高,可以提高双面粘合带的剪切粘合力。
在全部单体混合物中所占的丙烯酸2-乙基己酯的优选含量为10~40重量%。通过使丙烯酸2-乙基己酯的含量为10重量%以上,从而使上述丙烯酸类粘合剂层的粘合力提高,可以提高双面粘合带的剪切粘合力。通过使丙烯酸2-乙基己酯的含量为40重量%以下,从而使上述丙烯酸类粘合剂层处于适度的柔软度和内聚力,可以提高双面粘合带的剪切粘合力。
上述单体混合物可以根据需要包含除丙烯酸丁酯和丙烯酸2-乙基己酯以外的可共聚的其他聚合性单体。
作为上述可共聚的其他聚合性单体,可列举例如:(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸正丙酯、(甲基)丙烯酸异丙酯等烷基的碳数为1~3的(甲基)丙烯酸烷基酯;甲基丙烯酸十三烷基酯、(甲基)丙烯酸硬脂酯等烷基的碳数为13~18的(甲基)丙烯酸烷基酯;(甲基)丙烯酸羟基烷基酯、甘油二甲基丙烯酸酯、(甲基)丙烯酸缩水甘油酯、2-甲基丙烯酰基氧基乙基异氰酸酯、(甲基)丙烯酸、衣康酸、马来酸酐、巴豆酸、马来酸、富马酸等官能性单体。
为了将上述单体混合物共聚而得到上述丙烯酸类共聚物,只要使上述单体混合物在聚合引发剂的存在下进行自由基反应即可。作为使上述单体混合物进行自由基反应的方法、即聚合方法,使用现有公知的方法,可列举例如溶液聚合(沸点聚合或恒温聚合)、乳液聚合、悬浮聚合、本体聚合等。
上述聚合引发剂并无特别限定,可列举例如有机过氧化物、偶氮化合物等。作为上述有机过氧化物,可列举例如:1,1-双(叔己基过氧化)-3,3,5-三甲基环己烷、过氧化特戊酸叔己酯、过氧化特戊酸叔丁酯、2,5-二甲基-2,5-双(2-乙基己酰基过氧化)己烷、过氧化-2-乙基己酸叔己酯、过氧化-2-乙基己酸叔丁酯、过氧化异丁酸叔丁酯、过氧化-3,5,5-三甲基己酸叔丁酯、过氧化月桂酸叔丁酯等。作为上述偶氮化合物,可列举例如偶氮二异丁腈、偶氮双环己烷甲腈等。这些聚合引发剂可以单独使用,也可以并用2种以上。
上述丙烯酸类共聚物的重均分子量(mw)的优选的下限为40万、优选的上限为200万。通过使重均分子量为40万以上,从而上述丙烯酸类粘合剂层的内聚力提高,可以进一步提高双面粘合带的剪切粘合力。通过使重均分子量为200万以下,从而上述丙烯酸类粘合剂层的粘合力提高,可以进一步提高双面粘合带的剪切粘合力。重均分子量的更优选的下限为50万、更优选的上限为150万。
为了将重均分子量调整为上述范围,只要调整聚合引发剂、聚合温度等聚合条件即可。
需要说明的是,重均分子量(mw)是指基于gpc(gelpermeationchromatography:凝胶渗透色谱)得到的标准聚苯乙烯换算的重均分子量。
上述丙烯酸类粘合剂层可以含有增粘树脂。
作为上述增粘树脂,可列举例如松香酯系树脂、氢化松香系树脂、萜烯系树脂、萜烯酚系树脂、香豆酮茚系树脂、脂环族饱和烃系树脂、c5系石油树脂、c9系石油树脂、c5-c9共聚系石油树脂等。这些增粘树脂可以单独使用,也可以并用2种以上。
上述增粘树脂的含量并无特别限定,相对于上述丙烯酸类共聚物100重量份的优选的下限为10重量份、优选的上限为60重量份。通过使上述增粘树脂的含量为10重量份以上,从而上述丙烯酸类粘合剂层的粘合力提高,可以进一步提高双面粘合带的剪切粘合力。通过使上述增粘树脂的含量为60重量份以下,从而使上述丙烯酸类粘合剂层处于适度的硬度,粘合力或粘性提高,可以进一步提高双面粘合带的剪切粘合力。
上述丙烯酸类粘合剂层优选通过添加交联剂而在构成上述丙烯酸类粘合剂层的树脂(上述丙烯酸类共聚物和/或上述增粘树脂)的主链间形成交联结构。
上述交联剂并无特别限定,可列举例如异氰酸酯系交联剂、氮丙啶系交联剂、环氧系交联剂、金属螯合物型交联剂等。其中,优选异氰酸酯系交联剂。通过在上述丙烯酸类粘合剂层中添加异氰酸酯系交联剂,从而异氰酸酯系交联剂的异氰酸酯基与构成上述丙烯酸类粘合剂层的树脂中的醇性羟基反应,使上述丙烯酸类粘合剂层的交联变缓。因此,上述丙烯酸类粘合剂层可以使断续地施加的剥离应力分散,双面粘合带的剪切粘合力进一步提高。
上述交联剂的添加量相对于上述丙烯酸类共聚物100重量份优选为0.01~10重量份,更优选为0.1~3重量份。
上述丙烯酸类粘合剂层的交联度无论过高或过低,施加大的剪切方向的负荷时,均会有容易从被粘物剥离的情况,因此优选为5~40重量%,更优选为10~40重量%,特别优选为15~35重量%。
需要说明的是,丙烯酸类粘合剂层的交联度如以下所示地计算:采集丙烯酸类粘合剂层w1(g),将该丙烯酸类粘合剂层在乙酸乙酯中于23℃浸渍24小时,用200目的金属丝网过滤不溶解成分,对金属丝网上的残渣进行真空干燥,测定干燥残渣的重量w2(g),根据下述式(1)计算。
交联度(重量%)=100×w2/w1(1)
对于本发明的双面粘合带,双面粘合带的总厚度为80~150μm。本发明的双面粘合带虽然具有由发泡体构成的基材,但是即使使其很薄也能够兼顾冲击吸收性和再剥离性。双面粘合带的总厚度的优选的下限为90μm,优选的上限为100μm。
优选本发明的双面粘合带的md方向的拉伸强度为12n/10mm以上且td方向的拉伸强度为8n/10mm以上。
若本发明的双面粘合带的md方向及td方向的拉伸强度为上述下限以上,则在剥离双面粘合带时不会过度地伸展,因此可以提高双面粘合带的再剥离性。上述md方向的拉伸强度的更优选的下限为13n/10mm,上述td方向的拉伸强度的优选的下限为9n/10mm。
上述md方向的拉伸强度的上限并无特别限制,但优选的上限为25n/10mm,更优选的上限为20n/10mm。上述td方向的拉伸强度的上限并无特别限制,但优选的上限为15n/10mm,更优选的上限为12n/10mm。
需要说明的是,此时的伸长率并无特别限定,但优选为200%~500%左右。
需要说明的是,拉伸强度可以利用依据jisz0237的方法来测定。
上述拉伸强度可以通过调节上述基材的平均气泡直径以及上述丙烯酸类粘合剂层的交联度和剪切弹性模量来达成。
作为本发明的双面粘合带的制造方法,可列举例如如以下所示的方法。
首先,在丙烯酸类共聚物、增粘树脂、根据需要添加的交联剂等中加入溶剂,制作粘合剂a的溶液,将该粘合剂a的溶液涂布于基材的表面,将溶液中的溶剂完全干燥除去,形成丙烯酸类粘合剂层a。接着,在所形成的丙烯酸类粘合剂层a上将脱模膜以其脱模处理面与丙烯酸类粘合剂层a相对的状态进行重叠。
接着,除上述脱模膜之外另行准备脱模膜,在该脱模膜的脱模处理面涂布粘合剂b的溶液,将溶液中的溶剂完全干燥除去,由此制作在脱模膜的表面形成有丙烯酸类粘合剂层b的层叠膜。在形成有丙烯酸类粘合剂层a的基材的背面将所得的层叠膜以使丙烯酸类粘合剂层b与基材的背面相对的状态重叠来制作层叠体。然后,通过利用橡胶辊等对上述层叠体进行加压,从而可以得到在基材的双面具有丙烯酸类粘合剂层且丙烯酸类粘合剂层的表面被脱模膜覆盖的双面粘合带。
另外,也可以以同样的要领制作2组层叠膜,将这些层叠膜以使层叠膜的丙烯酸类粘合剂层与基材相对的状态重叠在基材的双面的各面上来制作层叠体,利用橡胶辊等对该层叠体进行加压,由此得到在基材的双面具有丙烯酸类粘合剂层且丙烯酸类粘合剂层的表面被脱模膜覆盖的双面粘合带。
本发明的双面粘合带的用途没有特别限定,优选被用于将电池组等构成便携电子设备的部件粘接固定于设备本体、被用于将汽车构件粘接固定于汽车本体(即被用于电子设备部件固定用途或车载部件固定用途)等。具体而言,在大型的便携电子设备中的部件的粘接固定、汽车构件(例如车载用面板)的粘接固定等中可以使用本发明的双面粘合带。
这些用途中的本发明的双面粘合带的形状并无特别限定,可列举长方形、框状、圆形、椭圆形、环型等。
发明效果
根据本发明,可以提供虽然为薄型但是冲击吸收性和再剥离性优异的双面粘合带。
附图说明
图1为表示双面粘合带的再剥离性的测定方法的示意图。
图2为表示在双面粘合带的落下冲击试验中使用的试验装置的示意图。
图3为表示双面粘合带的落下冲击试验的试验方法的示意图。
具体实施方式
以下列举实施例对本发明进行更详细地说明,但本发明并不仅限定于这些实施例。
(基材(a)的制造)
将作为聚烯烃系树脂的直链状低密度聚乙烯(埃克森化学公司制、商品名“exact3027”、密度:0.900g/cm3)100重量份、作为热分解型发泡剂的偶氮二甲酰胺3.5重量份、作为分解温度调节剂的氧化锌1.0重量份和作为抗氧化剂的2,6-二叔丁基对甲酚0.5重量份供给至挤出机,在130℃进行熔融混炼,挤出厚度约200μm的长条片状的聚烯烃系树脂组合物。
接着,对上述长条片状的聚烯烃系树脂组合物的双面照射4.5mrad加速电压500kv的电子射线而使其交联。接着,将交联后的聚烯烃系树脂组合物切割成400×400mm,用压制机加压至厚度达到约150μm。将加压后的聚烯烃系树脂组合物连续地送入利用热风和红外线加热器保持为250℃的发泡炉内,对其加热而使其发泡,并且一边使其发泡一边以md的拉伸倍率为1.3倍、td的拉伸倍率为2.0倍进行拉伸,由此得到厚度60μm的基材(a)。对所得的基材测定平均气泡直径、密度、发泡倍率。
(基材(b)~(d)和(g)的制造)
将聚烯烃系树脂组合物的配方变更为偶氮二甲酰胺1.5~5.0重量份,并将td的拉伸倍率调整为1.5倍~2.0倍,除这几点外,与基材(a)的制造同样地操作,得到基材(b)~(d)和(g)。对所得的基材测定平均气泡直径、密度、发泡倍率。
(基材(e)~(f)的制造)
将聚烯烃系树脂组合物的配方变更为偶氮二甲酰胺2.0~3.5重量份,并且连续地送入未利用压制机进行加压而利用热风和红外线加热器保持为250℃的发泡炉内,对其加热而使其发泡,将td的拉伸倍率调整为2.5倍~3.5倍,除这几点外,与基材(a)的制造同样地操作,得到基材(e)~(f)。对所得的基材测定平均气泡直径、密度、发泡倍率。
(粘合剂(a)的制备)
在具备温度计、搅拌机、冷凝管的反应器中加入丙烯酸丁酯75重量份、丙烯酸2-乙基己酯21重量份、丙烯酸3.8重量份、丙烯酸2-羟基乙酯0.2重量份和乙酸乙酯80重量份,进行氮气置换后,加热反应器来开始回流。接着,在上述反应器内添加作为聚合引发剂的偶氮二异丁腈0.1重量份。使其在70℃回流5小时,得到丙烯酸类共聚物(a)的溶液。对于所得的丙烯酸类共聚物(a),使用water公司制“2690separationsmodel”作为色谱柱,利用gpc法测定了重均分子量,结果为70万。
相对于所得的丙烯酸类共聚物(a)的溶液中所含的丙烯酸类共聚物(a)的固体成分100重量份,添加聚合松香酯15重量份、萜烯酚15重量份、氢化松香酯10重量份、乙酸乙酯(不二化学药品公司制)125重量份、异氰酸酯系交联剂(日本聚氨酯公司制商品名“coronatel45”)2.2重量份,进行搅拌,得到粘合剂(a)。
(粘合剂(b)的制备)
除了设定为丙烯酸丁酯62重量份、丙烯酸2-乙基己酯24重量份、丙烯酸乙酯10重量份以外,与丙烯酸类共聚物(a)同样地得到重均分子量为140万的丙烯酸类共聚物(b)的溶液。
除了使用所得的丙烯酸类共聚物(b)的溶液以外,与粘合剂(a)同样地得到粘合剂(b)。
(实施例1)
准备厚度150μm的脱模纸,在该脱模纸的脱模处理面涂布丙烯酸类粘合剂a,使其在100℃干燥5分钟,由此形成厚度20μm的丙烯酸类粘合剂层。将该丙烯酸类粘合剂层与表1所示的聚烯烃发泡体的表面贴合。接着,以同样的要领在该聚烯烃发泡体的相反的表面也贴合与上述相同的丙烯酸类粘合剂层。由此,得到被脱模纸覆盖的总厚度100μm的双面粘合带。
(实施例2~6、比较例1~5)
除了按照表中的记载来变更基材的种类和丙烯酸类粘合剂的种类、丙烯酸类粘合剂层的单面厚度以外,与实施例1同样地得到双面粘合带。
(拉伸强度和伸长率的测定)
将所得的双面粘合带切割成10mm×200mm的大小,制作测定用样品。接着,依据jisz0237,以5mm/sec的速度进行拉伸试验,计算拉伸强度(n/10mm)和伸长率(%)。
<评价>
对实施例、比较例中所得的双面粘合带进行了以下的评价。结果示于表1和表2。
(再剥离性)
图1中示出表示双面粘合带的再剥离性的测定方法的示意图。
首先,将双面粘合带切割成10mm×60mm。接着,在厚度2mm的聚碳酸酯(pc)板上粘贴所切割的双面粘合带。之后,如图1所示,将与粘贴有双面粘合带的pc板同样的pc板以使pc板的端部位于距离试验片的端部1cm的位置的方式进行贴合,以5kg压接10秒后,在23℃静置24小时,得到试验片。将静置后的试验片的双面粘合带一边拉伸一边剥离,将在中途未拉断地剥离的情况设为“○”,将在剥离的中途双面粘合带被拉断的情况设为“×”,评价了再剥离性。
(耐冲击性)
(1)试验装置的制作
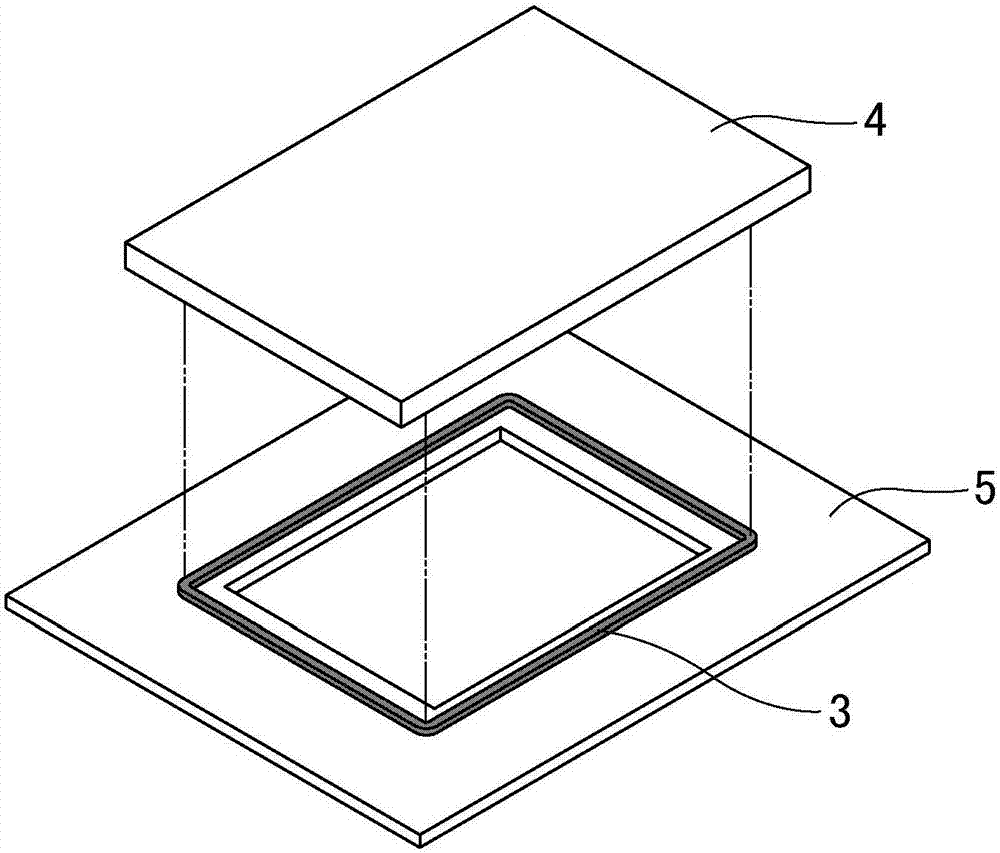
图2中示出在实施例、比较例中所得的双面粘合带的落下冲击试验中所使用的试验装置的示意图。
将所得的双面粘合带冲裁成外径为宽46mm、长61mm且内径为宽44mm、长59mm,制成宽1mm的框状试验片。接着,如图2所示,对于在中央部分空出宽38mm、长50mm的方形孔的厚度2mm的聚碳酸酯板5,将剥离脱模纸后的试验片3以使方形孔位于中央的方式进行粘贴后,从试验片3的上表面以使试验片3位于中央的方式粘贴宽50mm、长74mm、厚4mm的玻璃板4,从位于上表面的玻璃板4侧施加10秒5kgf的力,对于位于上下位置的玻璃板4和聚碳酸酯板5与试验片进行压接,在23℃放置24小时,制作试验装置。
(2)耐冲击性的评价
图3中示出实施例、比较例中所得的双面粘合带的落下冲击试验的试验方法的示意图。如图3所示,将所制作的试验装置翻转后固定于支撑台上,使可通过方形孔的大小的150g的铁制重物6以通过方形孔的方式落下。缓缓地提高落下铁制重物6的高度,对利用通过铁制重物6的落下所施加的冲击而剥离试验片3和玻璃板4时的铁制重物6的下落高度进行测量,将铁制重物6的下落高度为30cm以上的情况设为“○”,将不足30cm的情况设为“×”,评价了耐落下冲击性。
[表1]
[表2]
产业上的可利用性
根据本发明,可以提供虽然为薄型但是冲击吸收性和再剥离性优异的双面粘合带。
附图标记说明
1聚碳酸酯板
2双面粘合胶带
3试验片
4玻璃板
5聚碳酸酯板
6铁制重物
- 还没有人留言评论。精彩留言会获得点赞!