一种右丙亚胺的分析方法与流程
本发明属于药物分析
技术领域:
,具体来说,本发明涉及一种右丙亚胺有关物质的高效液相色谱分析方法。
背景技术:
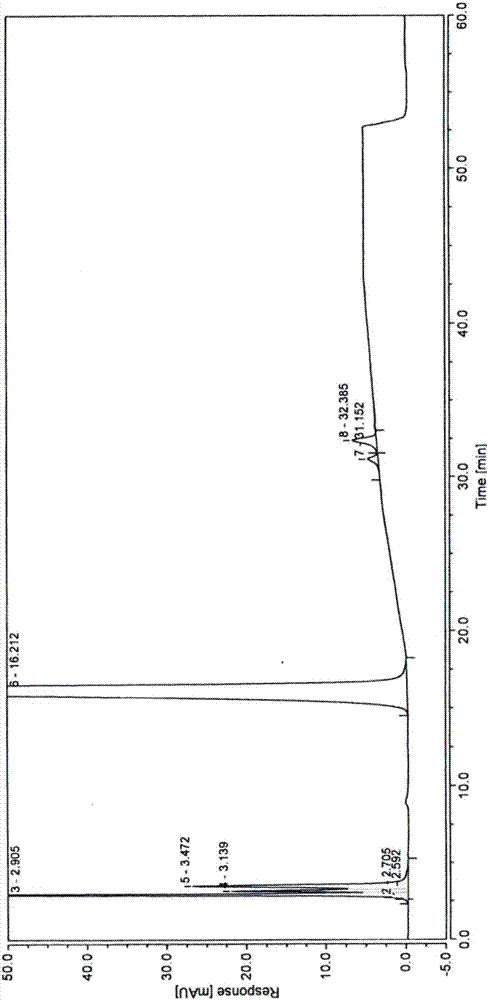
:右丙亚胺(Dexrazoxane)化学名为(S)-(+)-4,4’-(1-甲基-1,2-乙二基)-双-2,6-哌嗪二酮,其结构式为:右丙亚胺是丙亚胺(razoxane)的右旋异构体,又名右雷佐生、得拉唑沙或ICRF-187,是螯合剂乙二胺四乙酸的亲脂性衍生物,在临床上用作化学保护剂,主要用于预防蒽环类药物诱发的心脏毒性。蒽环类药物引起心脏毒性的机理主要是由于蒽环类药物与铁在形成稳定复合物过程中产生了活性氧,该活性氧对心肌膜发生脂质过氧化反应,损害心肌膜。右丙亚胺作为化疗药物的心脏保护剂,通过捕获自由铁和铁—蒽环类复合物中的铁,减少了对心肌组织产生毒性作用的氧自由基的生成而降低蒽环类药物心脏毒性。右丙亚胺由美国的Chiron公司开发,1992年首先在意大利上市,1995年FDA批准在美国上市。目前,右丙亚胺是临床上唯一用于减轻蒽环类抗肿瘤药物诱发心脏毒性的保护剂。专利WO2007/062076中披露了右丙亚胺有三种主要的降解杂质;专利文献CN201310326280.0公开了一种右丙亚胺冻干组合物杂质与含量的测定方法,其采用十八烷基硅烷键合硅胶为填充剂,以甲醇-0.01mol/L磷酸二氢钾溶液(15:85)为流动相。现有色谱分析技术对右丙亚胺原料药或制剂的杂质分离能力很弱,分离度不达标;杂质保留时间过短,无法达到分离目的;杂质检出数量很少,现有技术目前只能检测出3-4个杂质,与实际杂质数量差别很大。本领域亟需一种新的右丙亚胺的分析检测方法,使其能够有效、简便、快速地分析右丙亚胺原料药或其制剂中的有关物质,从而更好地控制产品质量。技术实现要素:为解决现有技术问题,本发明提供了一种新的右丙亚胺的分析方法,该分析方法采用梯度洗脱方式实现了杂质之间以及杂质与主峰之间的有效分离,适用于右丙亚胺原料药及其制剂(如冻干粉针及其他剂型)的有关物质分析。本发明主要采用了如下技术方案:一种右丙亚胺有关物质的高效液相色谱分析方法,所述分析方法采用耐纯水的低密度键合反相C18色谱柱;以流动相A和流动相B作为洗脱液进行梯度洗脱,所述流动相A为缓冲液,所述流动相B为有机溶剂;所述梯度洗脱的第一阶段中流动相A在所述洗脱液中的体积百分含量不低于90%,所述梯度洗脱的第一阶段的时长为15~30分钟。优选地,本发明中所采用的耐纯水的低密度键合反相C18色谱柱为WatersAtlantisT3或WatersACQUITYHSST3;更优选地,所述耐纯水的低密度键合反相C18色谱柱为WatersAtlantisT3;进一步优选地,所述WatersAtlantisT3色谱柱的规格为内径4.6mm,长度250mm,填料粒径5μm。本发明的分析方法采用梯度方式以流动相A和流动相B作为洗脱液进行洗脱,所述流动相A为缓冲液,所述流动相B为有机溶剂。优选地,所述缓冲液为选自磷酸二氢钾、磷酸二氢钠、磷酸氢二钠、磷酸氢二钾、磷酸氢二铵或磷酸二氢铵的缓冲盐溶液;更优选地,所述缓冲液为磷酸二氢钾或磷酸二氢钠溶液,进一步优选地,所述缓冲液为磷酸二氢钾溶液。优选地,所述缓冲液的浓度为1~50mmol/L,pH值为1~6;更优选地,所述缓冲液的浓度为5~15mmol/L,pH值为3.5~5.5;进一步优选地,所述缓冲液的浓度为8~12mmol/L,pH值为4.5~5。在本发明的一个具体实施方案中,所述缓冲液为10mmol/L、pH4.5~5的磷酸二氢钾溶液。优选地,所述有机溶剂选自甲醇、乙腈或异丙醇;更优选地,所述有机溶剂为甲醇或乙腈;进一步优选地,所述有机溶剂为甲醇。优选地,所述梯度洗脱的第一阶段中流动相A在所述洗脱液中的体积百分含量不低于95%,所述梯度洗脱的第一阶段的时长为15~20分钟。更优选地,所述梯度洗脱的第一阶段中流动相A在所述洗脱液中的体积百分含量不低于98%,所述梯度洗脱的第一阶段的时长为15~18分钟。在梯度洗脱的第一阶段,右丙亚胺三种主要降解杂质依次被洗脱出峰。本发明中的“第一阶段”是指梯度洗脱开始后的一段时间,例如从时间点0分钟至15分钟,从时间点0分钟到20分钟,从时间点0分钟到30分钟等。第一阶段的时长通常为15~30分钟,优选为15~20分钟。在本发明的一个具体实施方案中,第一阶段的时长为15分钟。优选地,所述梯度洗脱的条件具体为:其中10≤n≤90;优选地,50≤n≤80;进一步优选地,60≤n≤80。在本发明的一个具体实施方案中,n=70,亦即所述梯度洗脱的条件如下:优选地,在本发明的分析方法中,还具有如下任选的色谱条件:流动相流速为0.6~1.5ml/min,更优选为0.8~1.2ml/min,和/或色谱柱温度为5~20℃,优选为10~20℃,和/或检测器采用紫外检测器,检测波长为200~220nm,优选为203~213nm。在本发明的一个具体实施方案中,所述流动相流速为1.0ml/min;色谱柱温度为15℃;检测波长为208nm。本领域技术人员应知晓的是,除上述紫外检测器外,本发明的分析方法还可以灵活选用常用的其他类型高效液相色谱检测器,如荧光检测器、电化学检测器、示差检测器、蒸发光散射检测器、电导检测器、质谱检测器、电雾式检测器等。本发明的分析方法包括供试品溶液的制备、对照溶液的制备、系统适用性试验溶液的制备以及检测四个步骤。所述供试品溶液的制备包括:取右丙亚胺或含右丙亚胺的相关制剂适量,用稀释液配制成浓度为每1ml含0.5~2mg右丙亚胺的溶液,作为供试品溶液;所述对照溶液的制备包括:取供试品溶液,用稀释液稀释至100倍体积,作为对照溶液;所述系统适用性试验溶液的制备包括:取右丙亚胺或含右丙亚胺的相关制剂、杂质I对照品、杂质II对照品、杂质III对照品适量,加稀释液溶解并稀释制成每1ml含右丙亚胺0.5~2mg、杂质各5~20μg的溶液,作为系统适用性试验溶液;所述检测步骤包括:取系统适用性试验溶液10~20μl,注入液相色谱仪,右丙亚胺峰与相邻各杂质峰的分离度应符合要求;取对照溶液10~20μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的10~20%;精密量取供试品溶液10~20μl,注入液相色谱仪,记录色谱图;其中所述稀释液为盐酸溶液,各杂质的化学结构如下所示:优选地,本发明的分析方法包括如下步骤:(1)供试品溶液的制备:取右丙亚胺或含右丙亚胺的相关制剂适量,用盐酸溶液配制成浓度为每1ml含0.8~1.1mg右丙亚胺的溶液,作为供试品溶液;(2)对照溶液的制备:精密量取上述(1)制备的供试品溶液1ml,置100ml量瓶中,用(1)中所述盐酸溶液稀释至刻度,作为对照溶液;(3)系统适用性试验溶液的制备:取右丙亚胺或含右丙亚胺的相关制剂、杂质I对照品、杂质II对照品、杂质III对照品适量,加(1)中所述盐酸溶液溶解并稀释制成每1ml含右丙亚胺0.8~1.1mg、杂质各8~12μg的溶液,作为系统适用性试验溶液;(4)检测:取系统适用性试验溶液10μl,注入液相色谱仪,右丙亚胺峰与相邻各杂质峰的分离度应符合要求;取对照溶液10μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的10~20%;精密量取供试品溶液10μl,注入液相色谱仪,记录色谱图。在一个本发明的优选实施方案中,本发明的分析方法包括如下步骤:(1)供试品溶液的制备:取右丙亚胺或含右丙亚胺的相关制剂适量,用0.1mol/L盐酸溶液配制成浓度为每1ml约含1.0mg右丙亚胺的溶液,作为供试品溶液;(2)对照溶液的制备:精密量取上述(1)制备的供试品溶液1ml,置100ml量瓶中,用(1)中所述盐酸溶液稀释至刻度,作为对照溶液;(3)系统适用性试验溶液的制备:取右丙亚胺或含右丙亚胺的相关制剂、杂质I对照品、杂质II对照品、杂质III对照品适量,加(1)中所述盐酸溶液溶解并稀释制成每1ml含右丙亚胺约1mg、杂质各约10μg的溶液,作为系统适用性试验溶液;(4)检测:取系统适用性试验溶液10μl,注入液相色谱仪,右丙亚胺峰与相邻各杂质峰的分离度应符合要求;取对照溶液10μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的10%;精密量取供试品溶液10μl,注入液相色谱仪,记录色谱图。“约”在本发明中是指在所述数值的±10%以内。与现有技术相比,本发明的分析方法不但使得右丙亚胺主要降解杂质能够得到更好地保留与分离,杂质及主峰理论板数更高,可以检测出更多数量的杂质,而且方法学验证结果显示本发明方法重复性、灵敏度、耐用性、准确度均良好,为控制右丙亚胺原料药及其制剂的质量提供一种有效准确的检测方法。附图说明图1为对照例1条件下的色谱图;图2为对照例2条件下的色谱图;图3为对照例3条件下的色谱图;图4为对照例4条件下的色谱图;图5为实施例1条件下的系统适用性试验色谱图;图6为实施例1条件下的供试品溶液色谱图。具体实施方式下面结合具体实施例,对本发明进行进一步的描述。应理解的是,以下实施例仅用于进一步说明本发明,而不是对本发明范围的限制。本发明具体实施方式中使用的右丙亚胺(原料药)、右丙亚胺冻干制剂、杂质I对照品、杂质II对照品、杂质III对照品均来自江苏奥赛康药业股份有限公司;所使用的仪器为安捷伦1290高效液相色谱仪,包括G4220B1290BinPumpVL泵,G4212B1260DAD紫外检测器及ThermoChromeleon7.2色谱工作站。对照例11、色谱条件色谱柱:InertsilODS-SP色谱柱(150*4.6mm,5μm)流动相:甲醇-0.01mol/L磷酸二氢钾溶液(15:85)检测波长:208nm流速:1.0ml/min柱温:30℃2、实验步骤取右丙亚胺10mg,精密称定,加0.1mol/LNaOH溶液1.0ml,室温放置10min,加0.1mol/LHCl溶液中和,用0.01mol/L磷酸二氢钾溶液稀释制成每1ml中约含右丙亚胺1mg的溶液,作为待测溶液。【说明:对照例1-4中加入0.1mol/LNaOH溶液1.0ml,达到了碱破坏的效果,显著提高了待测溶液中杂质I、杂质II、杂质III的含量。】照高效液相色谱法(中国药典2005年版二部附录VD)并按上述条件进行测定。取待测溶液10μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20-30%,记录色谱图,结果见附图1及表1。由检测结果可以看出,杂质III、II、I色谱峰在此条件下重叠,右丙亚胺保留时间约为4.4min,杂质之间分离度不达标。该方法可以检测到样品中杂质数量不超过5个。表1对照例1条件下右丙亚胺与有关物质分离的试验结果注:“-”表示未检出。对照例21、色谱条件色谱柱:DikmaSpursilODS色谱柱(250*4.6mm,5μm)流动相:A为0.01mol/L磷酸二氢钠溶液B为甲醇检测波长:208nm流速:1.0ml/min柱温:30℃梯度洗脱条件:时间(分钟)流动相A(%)流动相B(%)09641596425901040851550851550.01964609642、实验步骤取右丙亚胺10mg,精密称定,加0.1mol/LNaOH溶液1.0ml,室温放置10min,加0.1mol/LHCl溶液中和,用0.01mol/L磷酸二氢钠溶液稀释制成每1ml中约含右丙亚胺1mg的溶液,作为待测溶液。照高效液相色谱法(中国药典2015年版四部附录0512)并按上述条件进行测定。取待测溶液10μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20-30%,记录色谱图,结果见附图2及表2。由检测结果可以看出,杂质III、II、I色谱峰在此条件下重叠,杂质III色谱峰与空白溶剂峰重叠,右丙亚胺保留时间约为16.2min,杂质之间分离度不达标。表2对照例2条件下右丙亚胺与有关物质分离的试验结果对照例31、色谱条件色谱柱:WatersXBridgeHilic色谱柱(250*4.6mm,5μm)流动相:A为0.01mol/L乙酸铵溶液B为乙腈检测波长:208nm流速:1.0ml/min柱温:30℃梯度洗脱条件:时间(分钟)流动相A(%)流动相B(%)059530505050505050.01595605952、实验步骤取右丙亚胺10mg,精密称定,加0.1mol/LNaOH溶液1.0ml,室温放置10min,加0.1mol/LHCl溶液中和,用0.01mol/L乙酸铵溶液稀释制成每1ml中约含右丙亚胺1mg的溶液,作为待测溶液。照高效液相色谱法(中国药典2015年版四部附录0512)并按上述条件进行测定。取待测溶液10μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20-30%,记录色谱图,结果见附图3。检测结果显示,主峰保留时间非常短并且分叉,杂质峰形不理想,基线不平整。该方法不能满足中国药典的要求。对照例41、色谱条件色谱柱:YMC-PackODS-AQ色谱柱(150*4.6mm,5μm)流动相:A为0.01mol/L磷酸二氢钾溶液B为甲醇检测波长:208nm流速:1.0ml/min柱温:20℃梯度洗脱条件:时间(分钟)流动相A(%)流动相B(%)01000510001096420901030901040802050802050.0110006010002、实验步骤取右丙亚胺10mg,精密称定,加0.1mol/LNaOH溶液1.0ml,室温放置10min,加0.1mol/LHCl溶液中和,用0.01mol/L磷酸二氢钾溶液稀释制成每1ml中约含右丙亚胺1mg的溶液,作为待测溶液。照高效液相色谱法(中国药典2015年版四部附录0512)并按上述条件进行测定。取待测溶液10μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20-30%,记录色谱图,结果见附图4及表3。检测结果显示,杂质III色谱峰与空白溶剂峰重叠,该方法不能满足中国药典的要求。表3对照例4条件下右丙亚胺与有关物质分离的试验结果注:“-”表示未检出或者未获得数据。实施例11、色谱条件色谱柱:WatersAtlantisT3色谱柱(250*4.6mm,5μm)流动相:A为10mmol/LKH2PO4溶液(pH4.5~5)B为甲醇检测波长:208nm流速:1.0ml/min柱温:15℃梯度洗脱条件:2、实验步骤取右丙亚胺冻干制剂,精密称定,加0.1mol/L盐酸溶液溶解并定量稀释制成每1ml中约含右丙亚胺1mg的溶液,作为供试品溶液。精密量取供试品溶液1ml,置100ml量瓶中,用0.1mol/L盐酸稀释至刻度,摇匀,作为对照溶液。另取右丙亚胺冻干制剂、杂质I、杂质II、杂质III对照品适量,加0.1mol/L盐酸溶液溶解并稀释制成每1ml中含右丙亚胺约1mg、杂质各约10μg的溶液,作为系统适用性试验溶液。照高效液相色谱法(中国药典2015年版四部附录0512)并按上述条件进行测定。取系统适用性试验溶液10μl,注入液相色谱仪,记录色谱图,结果见附图5及表4。由检测结果可以看出,杂质III、杂质II、杂质I、右丙亚胺依次出峰,保留时间分别为4.7min、7.2min、12.7min、34.9min,杂质之间以及杂质与主峰之间分离度均大于5,可以满足中国药典的要求。与对照例相比,本发明方法的杂质及主峰理论板数更高,可以检测到样品中杂质数量更多,一般在15~25个。表4实施例1条件下右丙亚胺与有关物质分离的系统适用性试验结果取对照溶液10μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的10%。精密量取供试品溶液、对照溶液各10μl,分别注入液相色谱仪,记录色谱图,其中供试品溶液的色谱图如附图6所示。检测结果显示,杂质III未检出,杂质II与I之间以及杂质与主峰之间均分离较好,可以满足中国药典的要求。实施例2~91、色谱条件实施例2~9的色谱条件中所采用的色谱柱和梯度洗脱条件与实施例1相同,其他色谱条件如下所示:2、实验步骤取右丙亚胺冻干制剂,按照上述实施例2~9色谱条件,并根据实施例1所述的实验步骤进行液相色谱分析,供试品溶液检测结果如表5所示:表5实施例2~9条件下右丙亚胺与有关物质分离的供试品溶液检测结果注:“-”表示未检出或者未获得数据。结果显示,在缓冲液浓度、波长、流速、柱温产生小幅变化时,主峰与杂质峰的分离度、理论板数均能符合要求,本发明方法的耐用性较好。另外,通过专属性、定量限与检测限、重复性、准确度等试验对本发明方法进行方法学验证性考察,结果显示本发明方法的专属性好,灵敏度高,重复性及准确度均良好,能够满足右丙亚胺原料药及其制剂分析检测的需要。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1