头花蓼不同浓度乙醇提取物的指纹图谱的测定方法与流程
本发明涉及中药领域,特别涉及中药质量评价领域,具体涉及一种热淋清颗粒及其原料药材头花蓼的质量评价方法,特别涉及热淋清颗粒及其原料药材头花蓼不同浓度乙醇提取物的指纹图谱的测定方法,更特别地具体涉及一种通过超高效液相色谱-飞行时间-质谱联用法(在本发明中可简称为UPLC-TOF-MS)对热淋清颗粒或药材头花蓼不同浓度乙醇提取物的指纹图谱的测定方法,以及对头花蓼进行质量评价的方法。头花蓼不同浓度乙醇提取物质量把控对于后续制剂的生产例如热淋清颗粒的生产具有重要意义。
背景技术:
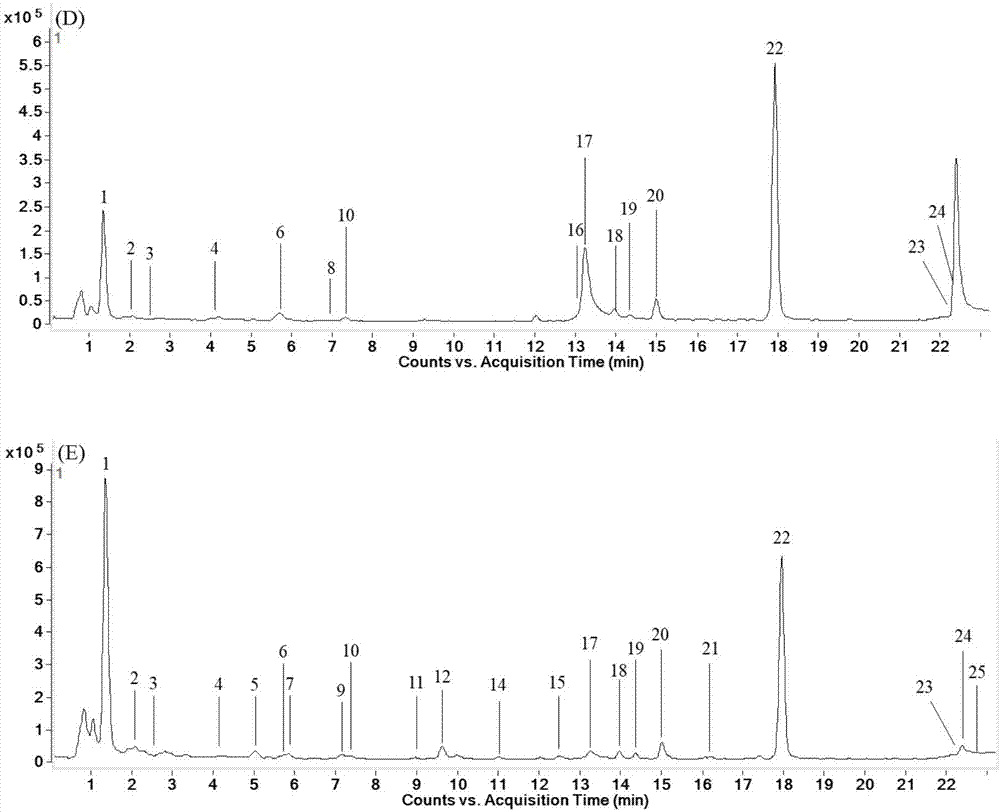
:头花蓼为蓼科植物头花蓼PolygonumcapitatumBuch.-Ham.exD.Don的干燥全草或地上部分,收载于《贵州省中药材民族药材质量标准》2003年版,具有清热利湿、抗菌消炎、利尿通淋等功效(贵州省食品药品监督管理局.贵州省中药材民族药材质量标准[M].贵阳:贵州科技出版社,2003:147)。头花蓼是贵州常用苗药,是贵州省中药现代化重点发展的“六大苗药”和重点培育发展的“七大中药产业链”的品种之一。本发明的申请人已在贵州施秉及贵阳乌当区建立了头花蓼规范化种植基地,并通过国家GAP认证。现在有关头花蓼GAP种植及化学成分等方面的研究已取得一定成果(孙长生,韩见宇,杨锦纲,等.头花蓼GAP种植基地的环境质量评价[J].中药研究与信息,2005,7(2):27~30),但头花蓼药材现行质量标准较为简单,仅以单一成分槲皮素为指标进行评价,无法达到真实、全面反应药材内在品质的目的。CN101091749A公开了一种头花蓼药材、提取物及其质量控制方法,其中的头花蓼药材具有如HPLC指纹图谱,含有13个特征峰,其中色谱条件为:色谱柱:依利特HYPERSILODS色谱柱(4.6mm×150mm,5μm);柱温:25℃;流动相:乙腈(A)-0.4%磷酸(B)溶液二元梯度洗脱,重量百分比为:0-30%:100-70%;流速:0.8mL/min;检测波长:310nm;进样量20μL。该发明质控制方法对不同产地头花蓼进行了考察比较,构建了头花蓼HPLC的指纹图谱。CN102296118A公开了一种DNA分子标记鉴别技术鉴别头花蓼与尼泊尔蓼的方法,在该发明中,以贵州具有代表性的民族药头花蓼为研究对象,探索使用RAPD分析方法对头花蓼与尼泊尔蓼进行遗传多样性分析;并在RAPD的实验基础上,将RAPD方法转化为SCAR标记,为头花蓼与尼泊尔蓼的药材鉴定提供了准确可靠、快速稳定的方法。DNA分子标记鉴别技术同样不适合药材的常规质量评价和控制。中药指纹图谱是指某种中药材或中成药中所共有的、具有特征性的某类或数类成分的色谱或光谱的图谱。在现阶段中药的有效成分绝大多数没有明确的情况下,中药指纹图谱对于有效控制中药材或中成药的质量具有重要的意义。日本汉方药主要生产企业在20世纪80年代就已经在企业内部采用高效液相指纹图谱控制质量。德国、法国在对银杏叶提取物联合开发的过程中,发现银杏叶提取物的医疗作用是提取物所得物质群的整体作用结果,而对这样一个整体的质量控制,亦采用高效液相指纹图谱方法。美国FDA最近几年制定的植物草药指南中已经明确把指纹图谱作为混合物质群的质量控制方法(FDA.GuidanceofIndusty:BotanicalDrug(Draft).2000August)。随着研究的深入,人们发现,作为中医理论的实践产物,中药,尤其是复方中药,其中所含的任一成分都不能代表其整体疗效。人们逐渐认识到,现行的参照西药(合成药)质量控制模式的质量标准不能恰当地反映中药内在的质量。从发展趋势看,从现行的质量控制模式向一种综合的、宏观的、可量化的鉴别与主要有效成分含量测定结合已是发展的趋势。中药质量评价的现行标准是利用光谱或色谱手段鉴别和测定某一种或几种有效成分、活性成分或指标成分,以及药典规定的常规检查项目。显然,这些质量标准的设置是模仿了化学药品的模式。其它国家如英国、印度、美国草药典、日本药局方中的汉药及德国CommissionE编辑的德国草药专论等也采用了基本相同的内容。对于化学药品而言,其药效成分为结构清晰的单一化合物,构效关系明确,其含量和纯度直接表达其有效及安全性。然而,中医用药的特点是复方配伍,任何单一的有效或活性成分的含量高低均不能表达其整体的疗效。例如,黄芪所含的黄芪甲苷(aastragalosideIV)是当前被选择为质量标准的鉴别和含量测定的最为常见的目标,但并没有依据证明黄芪甲苷与黄芪的功能主治的明确联系。同样,黄连、黄柏、三棵针均含小檗碱,一般都以它作为检测的目标,但是三者的功能主治却截然不同。复方制剂的情况就更加复杂。中医这种不是一对一的非线性的理论和实践说明中药质量应该采用某种宏观的综合的质量评价手段。头花蓼药材在制备其制剂例如热淋清颗粒过程中需要先进行提取,头花蓼不同浓度乙醇提取物质量把控对于后续制剂的生产例如热淋清颗粒的生产具有重要意义。本领域仍然需要有一从整体上控制头花蓼提取物的质量评价的方法,特别是可以用于头花蓼药材提取物的简便、宏观、可行的质量判定方法。技术实现要素:本发明的目的在于提供一种适合常规使用的用于热淋清颗粒或其原料药材头花蓼提取物质量评价的方法。本发明人发现采用一种独特的超高效液相色谱-飞行时间-质谱联用法(在本发明中可简称为UPLC-TOF-MS)可以有效地获得一个或多个方面的技术效果,该方法可以有效地为客观、准确地评价头花蓼不同乙醇浓度提取物的品质,为热淋清颗粒质量提升以及头花蓼药材提取工艺优化、规范生产工艺提供参考依据。本发明基于此发现而得以完成。为此,本发明第一方面提供了一种头花蓼提取物或包含该提取物的药物组合物的指纹图谱的测定方法,其是照超高效液相色谱-飞行时间-质谱联用法测定,包括以下步骤:(i)供试液配制:称取适量头花蓼提取物或包含该提取物的药物组合物粉末,加入甲醇水溶液制成混悬液,超声处理,过滤,必要时稀释,作为检测用试样;(ii)色谱及质谱分析条件:利用Agilent6230TOF-MS,MassHunter工作站采集一级质谱数据,ESI离子源,离子源温度:325℃,干燥气(氮气)流速:11L/min,雾化器气体(氮气)压力:40psi,鞘气(氮气)温度:350℃,鞘气流速:11L/min,毛细管电压:3500V,喷嘴电压:1500V,碎裂电压:175V,锥孔电压:65V,Oct1RFVpp:750V;负离子检测模式;扫描范围:100-1700amu;碰撞能量:10ev;以0.1%甲酸水溶液为流动相A,0.1%甲酸乙腈溶液为流动相B,洗脱程序为:在25-40分钟内使流动相A的比率从95%降到0%并且流动相B相应地从5%升高到100%;(iii)将步骤(i)试样注入液相色谱仪中,以步骤(ii)所述条件进行测定,获得供试样品的UPLC-TOF-MS总离子流色谱图。根据本发明第一方面的方法,其中步骤(i)中所述甲醇水溶液是浓度为60-80%的甲醇水溶液,例如约60%、约70%、约80%的甲醇水溶液。根据本发明第一方面的方法,其中步骤(ii)中所述流动相A是添加了0.02%异丙醇和0.0025%氨丁三醇的0.1%甲酸水溶液。根据本发明第一方面的方法,其中步骤(ii)中所述流动相B是添加了0.02%异丙醇和0.0025%氨丁三醇的0.1%甲酸乙腈溶液。根据本发明第一方面的方法,其中步骤(i)中所述混悬液的浓度为1-10mg/ml,例如2-10mg/ml,例如约2、5、10mg/ml。根据本发明第一方面的方法,其中步骤(i)中所述超声处理的时间为5-100min,例如10-60min,例如15-45min,例如15、30、45min。根据本发明第一方面的方法,其中步骤(i)中超声处理是以功率250W、频率33kHz的条件超声处理15-45min,例如约30min。根据本发明第一方面的方法,其中步骤(i)中所述稀释是使以提取物计的溶质在溶液中的浓度为0.1-1mg/ml,例如0.2-1mg/ml,例如0.3-1mg/ml,例如约0.2、0.5、1mg/ml。根据本发明第一方面的方法,其中步骤(i)中所述稀释使用的稀释液是浓度为60-80%的甲醇水溶液,例如约60%、约70%、约80%的甲醇水溶液。在一个实施方案中,步骤(i)中所述稀释使用的稀释液是步骤(i)中用于配制混悬液的甲醇水溶液。根据本发明第一方面的方法,其中使用的液相色谱仪是Waters公司的UPLC系统或者美国Agilent公司1290Infinity超高效液相色谱系统。根据本发明第一方面的方法,其中使用的飞行时间质谱是Bruker公司的Q-TOF-MS系统或者是Agilent6230TOF-MS系统。根据本发明第一方面的方法,其中步骤(i)中所述洗脱程序如下:时间(min)A(%)B(%)09550.59552081.518.528010030010030.195532955根据本发明第一方面的方法,其还包括对已知或符合规定的提取物或包含该提取物的药物组合物进行测定的步骤。根据本发明第一方面的方法,其还包括在步骤(iii)之后,将测试头花蓼提取物或包含该提取物的药物组合物的色谱图与已知或符合规定的头花蓼提取物或包含该提取物的药物组合物色谱图进行比较的步骤。在一个实施方案中,本发明第一方面的方法,其在步骤(iii)之后还包括如下步骤:将测试头花蓼提取物或包含该提取物的药物组合物的色谱图中各个峰面积大于主峰面积1%(例如大于2%)的色谱峰与已知或符合规定的头花蓼提取物或包含该提取物的药物组合物色谱图的相应色谱峰进行比较,比较二者保留时间的一致性。根据本发明第一方面的方法,其中测试头花蓼提取物或包含该提取物的药物组合物所指认的色谱峰及其对应物质包括:本发明任一方面或该任一方面的任一实施方案所具有的任一技术特征同样适用其它任一实施方案或其它任一方面的任一实施方案,只要它们不会相互矛盾,当然在相互之间适用时,必要的话可对相应特征作适当修饰。下面对本发明的各个方面和特点作进一步的描述。本发明所引述的所有文献,它们的全部内容通过引用并入本文,并且如果这些文献所表达的含义与本发明不一致时,以本发明的表述为准。此外,本发明使用的各种术语和短语具有本领域技术人员公知的一般含义,即便如此,本发明仍然希望在此对这些术语和短语作更详尽的说明和解释,提及的术语和短语如有与公知含义不一致的,以本发明所表述的含义为准。在本发明中,术语“药物组合物”或者“制剂”,它们在本发明中具有相同或类似含义,即均包括活性物质例如头花蓼提取物以及任选的药学可接受的辅料。在本发明中,术语“头花蓼药材”可以是头花蓼的干品或鲜品。在本发明中,术语头花蓼“提取物”可以是头花蓼药材经任何方法提取获得的提取物,例如溶剂(例如水、醇等)提取获得的,或者经超临界二氧化碳萃取获得的提取物。在本发明中,术语“头花蓼醇提取物”或者“头花蓼醇提浸膏”是指头花蓼用醇类溶剂(可以是含水或不含水的)或主要步骤用醇类溶剂提取所获得的提取物,所述醇类溶剂例如甲醇、乙醇等,例如其可以是如下方式获得:将头花蓼地上部分鲜品或干品加6-8倍量的5~90%乙醇回流提取1-3次,每次约1-5小时,过滤,使滤液浓缩、干燥,得醇提取物或醇浸膏。在本发明中,术语“头花蓼水提取物”是指头花蓼用水或主要步骤用水提取获得的提取物,例如参考中国药典2010年版一部“热淋清颗粒”项下的方法获得的头花蓼水提取物。尽管在本发明中头花蓼“提取物”可以是本发明提及的甚至是本发明未提及的,然而本发明作为一种检测方法,该方法的有效性并不会因为检测对象的改变而不能实施。恰恰相反,本发明方法一方面可以检测药材以及头花蓼药物制剂,另一方面还可以检测水提物,再一方面还可以检测醇提物,由此表明本发明适用场合非常广泛。附图说明图1:头花蓼30%(A)、50%(B)、70%(C)、95%(D)乙醇提取物与水提物(E)的UHPLC-TOF-MS的提取化合物色谱图(ECC)。图2:各色谱峰于头花蓼不同浓度乙醇提取物与水提物中的相对含量。具体实施方式通过下面的实施例可以对本发明进行进一步的描述,然而,本发明的范围并不限于下述实施例。本领域的专业人员能够理解,在不背离本发明的精神和范围的前提下,可以对本发明进行各种变化和修饰。本发明对试验中所使用到的材料以及试验方法进行一般性和/或具体的描述。虽然为实现本发明目的所使用的许多材料和操作方法是本领域公知的,但是本发明仍然在此作尽可能详细描述。实施例1:头花蓼药材、提取物及制剂UPLC-TOF-MS指纹图谱分析本实施例对本申请人提供的头花蓼不同浓度乙醇提取物或水提取物或者进一步的其制剂即热淋清制剂进行了UPLC-TOF-MS(Waters公司UPLC或美国Agilent公司1290Infinity超高效液相色谱系统;Bruker公司Q-TOF-MS,或Agilent6230TOF质谱系统)指纹图谱分析,鉴定了提取物和/或制剂中的主要成分。鉴定结果表明头花蓼提取物中主要含有黄酮及多酚类化合物。1、材料头花蓼30%、50%、70%、95%乙醇提取物、水提取物5个样品以及5个标准品(鞣花酸,没食子酸,槲皮苷,原儿茶酸,金丝桃苷)均由贵阳中医学院提供;头花蓼制剂热淋清,其基本上是由头花蓼提取物(70%乙醇提取物)加上适宜辅料制备得到的;甲醇﹑乙腈、甲酸为色谱纯(ACS公司),MilliQ水由Millipak-20FilterUnit处理,其他试剂均为分析纯。其中,以上5个提取物是照如下方法制备得到的:将头花蓼地上部分干品10Kg,加7倍量的提取溶剂(分别为30%乙醇、50%乙醇、70%乙醇、95%乙醇、水)回流提取2次,每次约1.5小时,过滤,使滤液浓缩、干燥,分别得提取物。2、样品液制备:称取适量头花蓼各浓度醇提取物或水提物粉末或热淋清颗粒,加入70%甲醇使浓度达5mg/ml,超声处理使尽量溶解,混悬液用70%甲醇稀释10倍,摇匀,滤过,续滤液过0.22μm微孔滤膜,备用于质谱检测。称取适量各标准品粉末,加入甲醇使浓度达20μg/ml,超声溶解,过0.22μm微孔滤膜,备用于质谱检测。3、色谱及质谱分析条件:美国Agilent公司1290Infinity超高效液相色谱系统。色谱柱:WatersACQUITYUPLCBEHC18色谱柱(2.1×100mm,1.7μm),柱温:40℃;流速:0.35ml/min;进样量:2μl。以添加了0.02%异丙醇和0.0025%氨丁三醇的0.1%甲酸水溶液为流动相A,以添加了0.02%异丙醇和0.0025%氨丁三醇的0.1%甲酸乙腈溶液为流动相B,按下表的规定进行梯度洗脱。UPLC-TOF-MS梯度洗脱程序时间(min)A(%)B(%)09550.59552081.518.528010030010030.195532955质谱条件:利用Agilent6230TOF-MS,MassHunter工作站采集一级质谱数据,ESI离子源,离子源温度:325℃,干燥气(氮气)流速:11L/min,雾化器气体(氮气)压力:40psi,鞘气(氮气)温度:350℃,鞘气流速:11L/min,毛细管电压:3500V,喷嘴电压:1500V,碎裂电压:175V,锥孔电压:65V,Oct1RFVpp:750V;负离子检测模式;扫描范围:100-1700amu。碰撞能量:10ev。4、数据分析首先根据现有研究结果(例如CN102809618A,中国专利申请号201210299950X记载的,其全部内容引入本文作为参考),利用从头花蓼醇提物与水提物中所鉴定出的53个化合物建立数据库。再根据一级质谱所提供的精确分子量﹑同位素丰度和同位素间距等信息与数据库中的化学成分进行匹配;同时利用与对照品比对出峰时间和精确分子量来确认部分色谱峰。最后获得各色谱峰的峰面积,并计算各色谱峰于各样品中的相对含量(峰面积/峰面积总和×100%),以评价不同浓度乙醇对提取物中化学成分的影响。5、结果5.1、头花蓼不同浓度乙醇提取物与水提物的化学成分鉴定按上文所述色谱及质谱条件进行检测,头花蓼不同浓度乙醇提取所得的提取物的色谱图见图1之(A)-(D),头花蓼水提物所得的提取化合物色谱图见图1之(E)。本实验利用出峰时间及精确分子量等讯息,把各色谱峰与已知头花蓼数据库中的化学成分及对照品进行匹配,获得鉴定结果。由于本实验所使用的数据库已经过前期研究的二级质谱验证,因此本实验并没有对所鉴定的色谱峰进一步进行二级质谱验证。采用上述分析手段,最后于不同浓度乙醇提取物与水提物中共鉴定出25个化合物,其鉴定结果见表1。从图1及表1可见,没食子酸(1)和槲皮苷(22)为头花蓼不同浓度乙醇提取物与水提物中的主要成分。其中95%乙醇提取物缺少部分trigalloylglucose(三没食子酰基葡萄糖)的同分异构体峰,这可能与该化合物的水溶性有关。表1:头花蓼不同浓度乙醇提取物与水提物UPLC-TOF-MS色谱图中指认的色谱峰*利用对照品所鉴定的色谱峰5.2、头花蓼不同浓度乙醇提取物与水提物化学成分对比分析头花蓼不同浓度乙醇提取物与水提物的化学成分对比图见图2。从图2可见,各化学成分于头花蓼不同浓度乙醇提取物与水提物中的含量具有明显的差异。当中可把各化学成分分为五类:第一类化学成分于样品中的含量随着样品中乙醇的浓度增加而减少(图2A),当中包括gallicacid(没食子酸,峰1),表示这一类成分的水溶性较高;第二类化学成分于样品中的含量随样品中乙醇浓度的先增加然后减少(图2B),这类化合物主要包括3,6-Digalloylglucose(峰2)和trigalloylglucose(峰4)及其同分异构体;第三类化合物于样品中的含量随样品中乙醇浓度的增加而增加(图2C),表示这类化合物的脂溶性比较高,这类化合物主要包括了davidiin(峰16)和ellagicacid(鞣花酸,17);第四类化合物的含量变化比较大,但它们含量的基本规律是随样品中乙醇含量的增加而增加(图2D),这类化合物主要包括了Protocatechuricacid(原儿茶酸,峰3)和quercitrin(槲皮苷,22);第五类化合物的含量在各乙醇浓度的样品中都没有太大的变化,一直保持比较平稳的状态(图2E),这类化合物主要包括了quercetin-3-O-β-D-glucopyranoside(峰18)和hyperoside(金丝桃苷,峰19)。另外,鞣花酸是头花蓼提取物中的一种重要物质。已经发现,鞣花酸在提取物中的含量与该提取物的提取溶剂有关,提取溶剂中乙醇浓度越高显示鞣花酸的含量越高。本发明实施例1的方法可以用于对头花蓼提取物中的典型物质进行定量,包括定量测定鞣花酸的含量。在对药剂中的活性物质进行含量测定时,需要考察方法学的回收率。一般而言,回收率与样品处理方法有关,而与后续仪器测定例如HPLC测定无关,对于一般化学药物或中药制剂中活性物质的含量测定而言,通常要求对各种活性物质的回收率达到95%以上特别是98%以上,本发明对于头花蓼提取物或其中药制剂即通常有这种要求;而对于一些生物样品,例如血浆中的化学物质含量测定,它们的回收率通常较低,通常没有特定要求,而是结合具体情形、具体药物确定,例如有时可以高达90%以上,有时低于50%亦可接受。这种回收率通常可以用于表征方法学的准确度。例如2015年版《中国药典》二部之“9101药品质量标准分析方法验证指导原则”中记载的“3.中药化学成分测定方法的准确度”一节,记载了回收率的测定方法,本发明测定回收率时即采用此方法。本发明实施例1方法经照上述“9101药品质量标准分析方法验证指导原则”,可以用于对各种提取物中的各种物质进行定量测定。已经测得五种提取物中鞣花酸的含量在0.36~6.33%范围内,提取溶剂乙醇浓度越高则鞣花酸的含量越高。另外,使用本发明实施例1的方法,已经测得鞣花酸、没食子酸、槲皮苷、原儿茶酸、金丝桃苷五者在各种提取物中的回收率均在96~102%范围内,并且对于同一化学物质,其在不同提取物中的回收率基本一致,具体地说,对于五种化学物质中的任一种,其在五种提取物中的回收率均相关不越过1.8%个百分点,例如,鞣花酸在五种提取物中的回收率均在98.2~99.6%范围内,显示具有优良的回收率。然而,本发明人在补充试验1中,发现如果在HPLC流动相组成作细微变化时,鞣花酸在五种不同提取物中的回收率呈现显著不同的分化,并且提取物中鞣花酸含量越高反而回收率越小。具体的补充试验1是这样进行的:除了流动相组成不同于实施例1外,其余均同实施例1进行操作;不同的流动相分三种情形:(a)仅是异丙醇和氨丁三醇二者在流动相A和B中均未添加、(b)仅是异丙醇在流动相A和B中均未添加、(c)仅是氨丁三醇在流动相A和B中均未添加;结果显示,(a)、(b)、(c)三种情形中鞣花酸的回收率分别在63.3~78.6%、82.7~89.4%、67.5~87.2%范围内,并且在(a)、(b)、(c)三种情形中提取物中的鞣花酸含量越高(即提取物所用提取溶剂乙醇浓度越高)反而回收率越小。按照常规理解,含量越低可预见的回收率可能会越低。本发明关于热淋清颗粒及其原料药材头花蓼不同浓度乙醇提取物的指纹图谱的测定方法,其是通过超高效液相色谱-飞行时间-质谱联用法对热淋清颗粒或药材头花蓼不同浓度乙醇提取物的指纹图谱的测定方法。通过本发明头花蓼不同浓度乙醇提取物质量把控,对于后续制剂的生产例如热淋清颗粒的生产具有重要意义。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1