一种测定黑臭水中邻苯二甲酸酯类化合物的方法与流程
本发明属于污染物测定
技术领域:
,具体地,涉及一种测定黑臭水中邻苯二甲酸酯类化合物的方法。
背景技术:
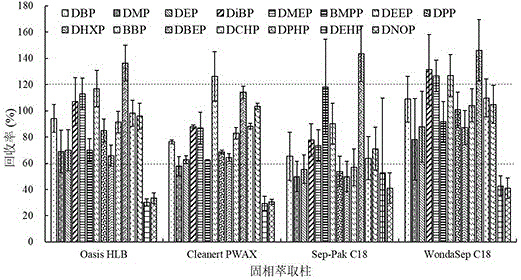
:随着城镇化和工业化的快速发展,部分城市河道沦为工业废水和生活污水排污的主要通道,随着污染物的增加水体的自净能力变弱,造成水体缺氧和富营养化,形成黑臭水体。水体黑臭的原因主要是有机污染物过量排入水体,使溶解氧降低,产生不同类型的黑臭类物质,呈现水体黑臭。目前城市黑臭水体已经成为一种城市病,几乎所有城市无一幸免。城市黑臭水体不仅给群众带来了极差的感官体验,而且影响群众生产生活导致严峻的水环境问题。国务院颁布的《水污染防治行动计划》提出“到2020年,地级及以上城市建成区黑臭水体均控制在10%以内,到2030年,城市建成区黑臭水体总体得到消除”的控制性目标。为加快城市黑臭水体整治,2015年住房城乡建设部会同环境保护部、水利部、农业部组织制定了《城市黑臭水体整治工作指南》,从此,我国城市地区全面开展整治城镇重污染河涌。整治黑臭水体,需先系统分析城市黑臭水体水质水量特征及污染物来源,并明确城市水体底泥中所含有的污染物,根据污染情况,按照“控源截污、内源治理;活水循环、清水补给;水质净化、生态修复”的总体要求,采取适当的整治技术。根据《城市黑臭水体整治工作指南》规定,城市黑臭水体分级的评价指标包括化学需氧量(cod)、氨氮(nh3-n)、溶解氧(do)等,这些指标都有成熟的现场快速测定方法。然而,影响水体黑臭的有机污染物种类多,特别是一些难降解有机污染物如邻苯二甲酸酯(paes)、多环芳烃(pahs)等,其中,以paes污染检出率高、污染程度重而成为困扰整治黑臭水体的一大难题。由于paes通常具有强内分泌干扰作用,其在水体中可严重影响水体及沉积物中的动物和微生物的群落结构和功能,对水生植物也具有一定毒性,影响水体的自净能力。因此,明确黑臭水体中paes种类及其污染特征,对提高水质净化和生态修复效果具有重要意义。目前,国内外建立了许多分析水体中邻苯二甲酸酯类化合物的可靠方法,其中,常见的萃取方法有液-液萃取法、固相萃取法和固相微萃取法等,常见的检测方法包括液相色谱-质谱联用法(lc-ms)、气相/液相色谱法以及酶联免疫法等。然而,有关黑臭水中有机污染物paes等的分析方法以及黑臭水的基质成分(cod、nh3-n等)对检测方法的性能影响(即基质效应)目前尚未见有报道。因此,亟需一种高效、灵敏且广泛适用于检测不同程度黑臭水中邻苯二甲酸酯化合物的高效分析方法。技术实现要素:本发明要解决的技术问题是克服现有技术的缺陷和不足,提供一种测定黑臭水中邻苯二甲酸酯类化合物的方法,以在萃取检测条件优化和基质标线的基础上建立一种高效、灵敏且适用于检测黑臭水中邻苯二甲酸酯类化合物的分析方法,从而明确黑臭水体中paes种类及其污染特征,提高水质净化和生态修复效果。本发明上述目的通过以下技术方案实现:一种萃取黑臭水中邻苯二甲酸酯类化合物的方法,该方法包括以下步骤:调节黑臭水水样ph至1~4后,采用wondasepc18柱或sep-pakc18柱进行固相萃取;将甲醇、二氯甲烷按体积比(1~4):(4~1)混合作为洗脱剂对上述萃取物进行洗脱,所述洗脱剂的用量为每1000ml黑臭水水样经过固相萃取后得到的萃取物总共加入洗脱剂4~10ml;将洗脱剂进行蒸发浓缩至尽干,溶剂溶解定容后得待测液备用。优选地,所述黑臭水体水样的体积为100~1000ml,以获得充足的样品进行试验检测。更优选地,所述黑臭水体水样的体积为1000ml。优选地,调节黑臭水水样ph至1~3,以在合适的ph范围内获得更好的萃取效率。优选地,调节黑臭水水样ph至3,以使目标化合物的萃取效率最佳。更优选地,利用盐酸或硫酸调节黑臭水水样ph。优选地,采用wondasepc18柱进行固相萃取,以实现最佳的萃取效果。优选地,将甲醇、二氯甲烷按体积比1:4混合作为洗脱剂。优选地,所述洗脱剂的洗脱次数为1~3次,以将黑臭水中的邻苯二甲酸酯类化合物洗脱完全。更优选地,所述洗脱剂的洗脱次数为2次,以达到合适的洗脱效果。优选地,所述洗脱剂洗脱的单次用量为1~5ml,以将黑臭水体中的邻苯二甲酸酯类化合物溶于洗脱剂中,方便后续检测。更优选地,所述洗脱剂洗脱的单次用量为3ml,以充分溶解黑臭水体中的邻苯二甲酸酯类化合物。优选地,所述黑臭水水样的cod值为40~1000mg/l,氨氮值为2~70mg/l。优选地,所述洗脱剂的蒸发浓缩温度为40~50℃。更优选地,所述蒸发方式为旋转蒸发以使容器受热均匀,进而使洗脱剂尽快蒸发浓缩。更优选地,所述容器为鸡心瓶。更优选地,所述洗脱剂的蒸发浓缩温度为40℃,以将邻苯二甲酸酯类化合物溶液浓缩,提高其检测灵敏度。优选地,用于溶解定容的溶剂为甲醇。进一步优选地,用甲醇溶解定容至0.5~2ml。更优选地,用甲醇定容后的体积为1ml。本发明还请求保护一种测定黑臭水中邻苯二甲酸酯类化合物的方法,将经过上述方法萃取后得到的待测液作为样品进行测定。优选地,上述测定黑臭水中邻苯二甲酸酯类化合物的方法包括以下步骤:s1.标准品的制备:以邻苯二甲酸酯为标样,采用黑臭水基质标线进行外标法定量,其中,邻苯二甲酸酯类的浓度范围为0.009~0.039μg/ml;s2.气相色谱-质谱分析:通过气相色谱-质谱联用仪对上述萃取得到的待测液和s1所述的标准品分别进行分析检测,得到相关色谱图。优选地,步骤s2中,所述气相色谱-质谱联用仪毛细管色谱柱为石英毛细管色谱柱hp-5ms、hp-1ms或db-5ms中的一种或多种。更优选地,所述气相色谱-质谱条件为:采用毛细管色谱柱,固定相为hp-5ms,规格为30m×0.25mm×0.1μm;进样口温度为280~300℃;柱流量为1.0~1.5ml/min;进样量为1~3μl,不分流;程序升温步骤为:100℃保留2min,以15℃/min升至129℃,再以40℃/min升至280℃,保留5min。优选地,固定相hp-5ms通过5%二苯基与95%二甲基聚硅氧烷混合得到。优选地,所述邻苯二甲酸酯类标准品的制备方法具体为:准确称取各目标邻苯二甲酸酯类化合物标准品,以色谱纯甲醇定容,获得100μg/ml标准储备液,分别采用甲醇稀释标准储备液获得系列目标化合物标准品,标准品的浓度范围为0.05~2μg/ml。用气相色谱质谱联用仪进行测定时,以平行操作的邻苯二甲酸酯类标准品进行比较,以保留时间和优化的特征离子进行定性,以基质背景干扰少,峰形好、信噪高的特征离子为定量离子,以基质标线外标法进行定量。优选地,所述气相色谱-质谱联用仪的质谱条件为:ei源温度250℃,检测电压1.1kv,采用选择离子模式检测(sim)。更优选地,测定样品后以邻苯二甲酸酯标样的基质标线进行外标法定量。该方法能够检测出黑臭水中15种邻苯二甲酸酯类化合物,所述邻苯二甲酸酯类化合物分别为邻苯二甲酸二甲酯(dmp)、邻苯二甲酸二乙酯(dep)、邻苯二甲酸二异丁酯(dibp)、邻苯二甲酸二丁酯(dbp)、邻苯二甲酸二(2-甲氧基)乙酯(dmep)、邻苯二甲酸二(4-甲基-2-戊基)酯(bmpp)、邻苯二甲酸二(2-乙氧基)乙酯(deep)、邻苯二甲酸二戊酯(dpp)、邻苯二甲酸二己酯(dhxp)、邻苯二甲酸丁基苄基酯(bbp)、邻苯二甲酸二(2-丁氧基)乙酯(dbep)、邻苯二甲酸二环己酯(dchp)、邻苯二甲酸二(2-乙基)己酯(dehp)、邻苯二甲酸二正辛酯(dnop)、邻苯二甲酸二(2-丙基庚)酯(dphp)。优选地,上述方法可用于研究黑臭水中邻苯二甲酸酯类化合物含量水平、污染特征及风险评价。现有技术所公开的水样大多为普通自然水样,其cod值通常在30以下,氨氮值在1.5以下,而本发明是专门针对城市黑臭水体水样,即劣五类水设计的检测方法,发明人通过创造性的劳动将cod值为40~1000mg/l,氨氮值为2~70mg/l的黑臭水调节ph至1~4后,采用wondasepc18柱或sep-pakc18柱进行固相萃取,以甲醇和二氯甲烷的混合溶液为洗脱剂,并利用气相色谱-质谱联用仪对黑臭水中的邻苯二甲酸酯类化合物组分进行定量,有效提高了目标paes测定的准确度和紧密度。与现有技术相比,本发明具有以下有益效果:本发明所述方法适用范围广,对cod和氨氮变化范围大的黑臭水样品均能够高效检测,而且能够同时测定黑臭水中的15种邻苯二甲酸酯类化合物,检出限为0.009~0.039μg/l,具有回收率高、精密度好、灵敏度高、抗基质成分干扰的优点。附图说明图1为不同萃取柱对目标邻苯二甲酸酯化合物的萃取回收率;图2为溶液ph值对目标邻苯二甲酸酯化合物萃取回收率的影响。图3为不同洗脱剂对目标邻苯二甲酸酯化合物洗脱效率的影响。图4为不同洗脱剂用量对目标邻苯二甲酸酯化合物回收率的影响。图5为黑臭水基质样品中目标邻苯二甲酸酯化合物(1μg/l)的色谱图。图1~5中,dbp代表邻苯二甲酸二丁酯;dmp代表邻苯二甲酸二甲酯;dep代表邻苯二甲酸二乙酯;dibp代表邻苯二甲酸二异丁酯;dmep代表邻苯二甲酸二(2-甲氧基)乙酯;bmpp代表邻苯二甲酸二(4-甲基-2-戊基)酯;deep代表邻苯二甲酸二(2-乙氧基)乙酯;dpp代表邻苯二甲酸二戊酯;dhxp代表邻苯二甲酸二己酯;bbp代表邻苯二甲酸丁基苄基酯;dbep代表邻苯二甲酸二(2-丁氧基)乙酯;dchp代表邻苯二甲酸二环己酯;dphp代表邻苯二甲酸二(2-丙基庚)酯;dehp代表邻苯二甲酸二(2-乙基)己酯;dnop代表邻苯二甲酸二正辛酯。具体实施方式以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本
技术领域:
常规试剂、方法和设备。除非特别说明,以下实施例所用试剂和材料均为市购。i级黑臭水样(cod为160mg/l;氨氮为23.1mg/l)、ii级黑臭水样(cod356mg/l,氨氮70.7mg/l)、iii级黑臭水样(cod为1000mg/l;氨氮为73.6mg/l)。实施例1黑臭水中邻苯二甲酸酯化合物前处理方法的优化一、方法1、实验目的以实验室配置的ii级黑臭水样(cod356mg/l,氨氮70.7mg/l)为目标水样,通过加标回收率实验(各paes浓度均为1μg/l)探究固相萃取柱种类、水样ph值、洗脱剂种类及用量对黑臭水中paes化合物回收率的影响,从而获得优化的前处理方法。发明人对固相萃取柱、黑臭水样的ph值、洗脱剂种类、洗脱剂用量四个因素进行优化。其中,固相萃取柱分别选用oasishlb(waters,美国)、pwax(博纳艾杰尔,天津)、wondasepc18(shimadzu,日本)和sep-pakc18(waters,美国);ph值分别为1、2、3、4、5、6、7;洗脱剂分别为甲醇、二氯甲烷、甲醇二氯甲烷混合溶液(2:8,v/v)、甲醇二氯甲烷混合溶液(3:7,v/v)、甲醇二氯甲烷混合溶液(5:5,v/v)、甲醇二氯甲烷混合溶液(7:3,v/v)、甲醇二氯甲烷混合溶液(8:2,v/v);洗脱剂用量分别为为2ml、4ml、6ml、8ml、10ml。2、回收率实验过程(1)加标样品的制备:以甲醇为溶剂,将paes混合标准液(10mg/l)进行稀释,获得paes标准工作液(1mg/l),然后取一定量paes标准工作液,以ii级黑臭水样品稀释,获得含1μg/lpaes的污染黑臭水。(2)样品萃取:取1000mlpaes污染黑臭水水样,用盐酸调节ph值至3,用wondasepc18(shimadzu,日本)固相萃取柱,分别取3ml甲醇和二氯甲烷溶液作为洗脱剂用固相萃取柱洗脱,收集洗脱剂于鸡心瓶,40℃下旋转蒸发至尽干,用甲醇溶解定容为1ml,待测。(3)样品测定:用气相色谱-质谱联用仪进行测定,并用基质标线外标法进行定量分析,回收率通过加标样品测定浓度与其加标浓度比较获得,即:回收率=样品测定浓度/样品加标浓度×100%。测定过程中色谱条件为:hp-5ms石英毛细管色谱柱(30m×0.25mm×0.1μm),进样口温度280℃,载气为高纯氦气(≥99.999%),柱流量1.0ml/min,不分流进样(1μl);程序升温条件为:100℃保留2min,以15℃/min升至129℃,再以40℃/min升至280℃,保留5min;质谱条件为:ei源温度250℃,检测电压1.1kv,采用选择离子模式检测(sim),其他实验条件的操作均与该条件下的相同,只是固相萃取柱种类、水样ph值、洗脱剂种类及用量发生变化。二、结果(1)固相萃取柱种类的优化实验结果如图1所示,从图1可以看出,当以pwax柱和sep-pakc18柱萃取时,多数paes化合物的回收率≤60%,且部分化合物如deep和dehp标准偏差较大(>30%);以oasishlb萃取时,paes化合物回收率总体在60%~120%之间,但dehp和dnop回收率低于40%;以wondasepc18萃取时,各paes化合物的回收率总体在80%~120%之间,且dehp和dnop回收率在50%左右,优于其他固相萃取柱。由此选定wondasepc18为萃取柱。(2)ph值的优化实验结果如图2所示,从图2可以看出,弱酸性和中性条件下,目标paes回收率较低,其中ph值为5或7时,大部分的paes回收率低于40%;这与该条件下paes易于水解流失有关。随着ph值的降低,目标paes的回收率大幅提高,尤其ph值为1或3时目标化合物萃取效率最优,大部分目标化合物的回收率在80%~120%之间,但各paes回收率的标准偏差普遍较大(~20%),且其dehp和dnop的回收率显著低于ph值为3时。因此,选择将黑臭水样品ph值调至3。(3)洗脱剂的优化将黑臭水样品经洗脱剂洗脱后,所得15种paes化合物的回收率如图3所示,由图3可知,以甲醇为洗脱剂时,各目标化合物回收率较低,普遍在60%以下,甚至部分化合物回收率只有20%左右,这说明paes化合物的回收率与甲醇具有较强极性有关;以二氯甲烷为洗脱剂时,目标化合物回收率较甲醇洗脱剂时有所提高,但dphp、dehp、dnop等化合物回收率较低(<60%);以甲醇和二氯甲烷混合溶剂为洗脱剂时,目标化合物的回收率较单一溶剂时有显著改善,其中以二者配比为2:8和5:5时各paes回收率较好,然而由于dehp和dnop的回收率在甲醇和二氯甲烷混合溶剂配比为2:8(即1:4)时更优,故选择将甲醇和二氯甲烷按体积比1:4进行混合作为洗脱剂。(4)洗脱剂用量的优化从图4可以看出不同洗脱剂用量对目标paes化合物回收率的影响,当洗脱剂体积为2ml(即甲醇和二氯甲烷各1ml)时,目标paes的回收率普遍在60%以下,难以达到满意的萃取要求;随着洗脱剂体积的增加,各目标化合物的回收率显著提高,当其体积为4(即甲醇和二氯甲烷各2ml)或6ml(即甲醇和二氯甲烷各3ml)时,二者对目标paes提取的回收率相当,总体在80%~120%以内,但后者dphp和dnop的回收率较差(低于60%),显著低于前者(65%~77%);继续增加洗脱剂体积至8ml(即甲醇和二氯甲烷各4ml)或10ml(即甲醇和二氯甲烷各5ml)未能进一步增加目标paes的回收率,反而造成部分化合物回收率过高(dibp、dmep和deep回收率>120%)或过低的现象(dnop<60%),这可能与共萃取基质成分造成的基质效应有关。由此可见,洗脱剂为6ml时,目标paes回收率最优,故选其为洗脱剂体积。因此,测定黑臭水中paes化合的主要前处理方法为:用wondasepc18小柱萃取,调节黑臭水样品ph值至3,再将3ml甲醇和3ml二氯甲烷混合所得的混合液作为洗脱剂。实施例2优化前处理方法对不同程度黑臭水的适用性验证一、实验目的在优化的前处理条件下,测定不同程度黑臭水(cod为160~1000mg/l;氨氮为23.1~73.6mg/l)中不同浓度(0.2、1、2μg/l)目标paes的回收率和标准偏差。二、方法1、前处理其前处理方法与实施例1所得的最佳处理条件相同。2、样品制备以去离子水为母液,以少量无污染清澈湖水为微生物媒介,以敞口静置培养方式,通过调节碳源、氮源及微量元素等的配比,制备获得不同程度黑臭水基质水样(cod为160~1000mg/l;氨氮为23.1~73.6mg/l),其中黑臭水级别越高,其cod含量越大。用配置的不同级别黑臭水稀释paes(1mg/l)工作液制备获得含目标paes(0.2、1、2μg/l)的污染黑臭水,备用。3、不同级别黑臭水基质paes化合物标准曲线的配置分别取未污染的不同级别黑臭水样品,经实施例1所述最佳条件前处理后获得各级别黑臭水的萃取基质,之后分别以各黑臭水基质稀释目标paes工作液(10mg/l),获得各黑臭水的系列基质加标标样(0.05、0.1、0.2、0.5、1、2、5μg/ml),后以气相色谱质谱联用仪测定,以外标法进行定量分析,并通过线性回归分析获得各黑臭水的基质标线和检出限。4、各目标paes化合物选择性验证分别取出不同程度黑臭水基质样品,经实施例1所述最佳条件前处理后获得萃取基质,之后分别以不同程度黑臭水稀释目标paes化合物标准工作液(1mg/l),获得各黑臭水的基质加标样(1μg/l),然后用气相色谱质谱联用仪进行色谱分离、测定,根据所得色谱图中各目标paes化合物的分离度及其受基质杂峰干扰的程度判断本方法的选择性。5、各黑臭水样目标paes回收率及精密度验证分别取出不同级别黑臭水样品,向其中分别加入paes标准工作液(1mg/l),获得各黑臭水不同浓度加标样品(0.2、1、2μg/g),经实施例1所述最佳条件前处理后采用气相色谱质谱联用仪进行色谱分测定,并以基质标线外标法进行定量分析,回收率则以加标样品测定浓度与其加标浓度比较获得,精密度则由同一浓度加标样品测定值(同一浓度设置5个平行样)的相对标准偏差表示。三、结果实验结果如图5和表1~3所示,由表1可以看出,各级别黑臭水中目标paes化合物在0.05~2mg/l均表现出满意的线性(r2>0.99)。目标paes化合物在不同级别黑臭水中的仪器检出限为0.01~0.04mg/l,在100倍浓缩条件下(取100ml水样,浓缩至1ml检测)的方法检出限为:0.09~0.44μg/l(i级黑臭水)、0.10~0.39μg/l(ii级黑臭水)及0.12~0.38μg/l(iii级黑臭水),表现出高灵敏度。表1不同级别黑臭水样中邻苯二甲酸酯标线及检出限注:1)线性范围单位为mg/l;2)检出限单位为μg/l从图5可以看出,各目标paes化合物在混标样(0.5μg/ml)及各黑臭水基质标样(0.5μg/ml)均可根据其保留时间(7.78~30.66min)和特征离子(m/z)有效分离如下表2,且在各基质空白加标样中未检出对目标paes具有干扰的色谱峰,可见本发明提供方法具有较高的分离度和选择性。表2各paes化合物色谱保留时间和质谱特征离子各黑臭水基质中目标paes化合物不同浓度(0.2、1、2μg)加标样品的回收率总体在70%~120%之间,标准偏差(rsd)低于16%,如下表3所示。部分化合物回收率相对较低(55%~60%)与黑臭水基质成分复杂性及多组分痕量有机污染物性质的差异性的差异性有关。可见,本发明提供的方法具有较高的准确性和精密度。表3邻苯二甲酸酯化合物在不同程度黑臭水中的回收率和相对标准偏差实施例3建立方法对实际样品的测定一、实验目的为验证本发明方法的可行性,以此方法测定实际黑臭水中目标paes的含量。二、方法采集实际样品10个,均为广州市黑臭河涌水样,其水质常年为劣五类,cod和氨氮含量通常高于100mg/l和10mg/l。按照“实施例1”和“实施例2”的方法对样品进行分析测定,测定的样品包括实际样品及其加标质控样(1000ng/l)。三、结果测定结果如表4和表5所示,实际样品的加标回收率为65~115.8%,相对标准偏差低于18.0%;各实际样品中均全部检出15种目标paes化合物,其总浓度为2588~20459ng/l,以dphp检出量最高(660~12909ng/l),其次为dehp(234~6013ng/l)、dibp(195~863ng/l)和dbp(183~483ng/l),其余化合物检出量较低(主要在0.1~50ng/l)。本实施例显示了本发明所述方法具有较高的可行性和实际适用性。表4实际黑臭水中目标邻苯二甲酸酯化合物的检出浓度(ng/l)采样点dbpdmpdepdibpdmepbmppdeepdpp采样点146439468632725467.6采样点2356496356315861.4采样点3483518464012990.3采样点43496614791304170.5采样点51837771951242590.9采样点625691573321412971.2采样点7233658254155591.1采样点838811444332161771.5采样点92701118627316817141.2采样点1022187026114849130.3表5实际黑臭水中目标邻苯二甲酸酯化合物的检出浓度(ng/l)采样点bbpdbepdchpdphpdehpdnop总含量采样点1235313660297172588采样点2102070.266423122211045采样点326230.15132340.92097采样点481170.1671631771.911092采样点52670.2532525040.68484采样点66190.12574527130.99417采样点7231460.04870741090.213706采样点8241610.081290960130.920459采样点920240.0475913566212144采样点103200.1708033150.611189综上所述,本发明提供了一种可同时测定黑臭水样中15种paes化合物的高效分析方法。该方法以wondasepc18小柱进行固相萃取,以甲醇-二氯甲烷混合溶液进行洗脱,以气相色谱-质谱联用仪进行测定,并以黑臭水基质配置系列梯度标准曲线,以外标法定量。该方法具有可靠、精密、高灵敏度和抗复杂基质干扰的特征,可为开展水环境系统中邻苯二甲酸酯化合物污染特征和风险水平的研究提供可靠分析方法。实施例4测定黑臭水中邻苯二甲酸酯化合物的方法一、方法一种测定黑臭水中邻苯二甲酸酯化合物的方法,包括如下步骤:(1)样品前处理:取1000ml黑臭水水样,以盐酸(hcl)调节其ph值至3后,以wondasepc18小柱进行固相萃取,以甲醇和二氯甲烷按体积比1:4的混合溶液作为洗脱剂进行2次洗脱,每次洗脱剂用量为3ml,收集洗脱剂于鸡心瓶40℃旋转蒸发至尽干,用甲醇溶解定容为1ml后待测;(2)样品测定:采用气相色谱质谱联用仪进行测定,采用的色谱柱为hp-5ms石英毛细管色谱柱(30m×0.25mm×0.1μm),进样口温度280℃,柱流量1.0ml/min,不分流进样(1μl),程序升温步骤为:100℃保留2min,以15℃/min升至129℃,再以40℃/min升至280℃,保留5min;质谱条件为ei源温度250℃,检测电压1.1kv,采用选择离子模式检测(sim)测定样品后以邻苯二甲酸酯标样的基质标线进行外标法定量。二、结果通过本发明所述方法测得所述邻苯二甲酸酯类化合物分别为邻苯二甲酸二甲酯(dmp)、邻苯二甲酸二乙酯(dep)、邻苯二甲酸二异丁酯(dibp)、邻苯二甲酸二丁酯(dbp)、邻苯二甲酸二(2-甲氧基)乙酯(dmep)、邻苯二甲酸二(4-甲基-2-戊基)酯(bmpp)、邻苯二甲酸二(2-乙氧基)乙酯(deep)、邻苯二甲酸二戊酯(dpp)、邻苯二甲酸二己酯(dhxp)、邻苯二甲酸丁基苄基酯(bbp)、邻苯二甲酸二(2-丁氧基)乙酯(dbep)、邻苯二甲酸二环己酯(dchp)、邻苯二甲酸二(2-乙基)己酯(dehp)、邻苯二甲酸二正辛酯(dnop)、邻苯二甲酸二(2-丙基庚)酯(dphp),说明本发明所述方法适用范围广,对cod和氨氮变化范围大的黑臭水样品均能够高效检测,而且能够同时测定黑臭水中的15种邻苯二甲酸酯化合物,检出限为0.009~0.039μg/l,具有回收率高、精密度好、灵敏度高、抗基质成分干扰的优点。上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1