利用二维液相分离制备抗体类产品相关杂质的方法与流程
本发明涉及一种利用二维液相分离制备抗体类产品相关杂质的方法,属于抗体类产品分析
技术领域:
。
背景技术:
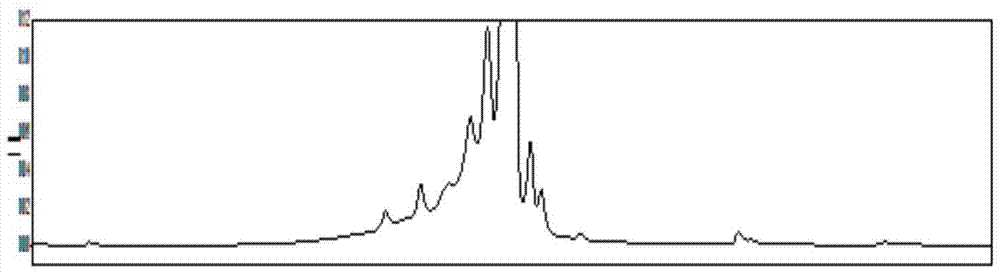
:单抗类免疫治疗药物在生产、运输、储存、销售、使用的过程中都会发生修饰或降解,产生杂质,这些杂质均有可能会对产品活性、质量产生影响,因此需要对药物中杂质做充分的理化性质、生物活性、药理毒理研究,以求了解它们的性质,以便在生产工艺中控制它们的生成,运输储存过程中防止它们的产生,从而确保药品的质量从工厂到患者或消费者都始终能达到质量标准,另外,从发展趋势看,由于市场及监管部门对药品品质提出越来越高的要求,越来越细微的抗体类产品相关杂质都被要求进行研究,因此,需要大量杂质作为样品来进行多项研究。现阶段,在抗体类产品研发及生产企业中,想要得到大量的抗体类产品相关杂质样品,必须在hplc分析后使用人工手动或是使用在线馏分收集器从hplc废液端收集杂质馏分,由于通常收集到的杂质馏分浓度非常低,无法利用,而且含有大量前端色谱流动相所含有的不利于杂质样品保存的物质,因此,必须将样品通过超滤、透析等方法置换到适合杂质样品保存的缓冲液当中,然后将其浓缩至可以用来分析检测的浓度,此过程中大量样品会因吸附在器具表面而损失,导致回收率非常低,通常在10%以下,在如此低回收率的情况下,想要得到足够多的样品去完成杂质完整分子量的测试,通常需要长达十几个工作日的反复收集,效率非常之低下,伴随着大量人力物力的消耗,另外,由于过程冗长,收集到的杂质可能发生了二次降解或修饰,使收集到的杂质纯度较低。技术实现要素:为了解决上述现有技术中存在的技术问题,本发明的目的在于提供一种利用二维液相分离制备抗体类产品相关杂质的方法,提高了各种抗体类相关杂质分离制备的回收率,较大的提高了效率,节省了人力,且提高了分离制备的杂质的纯度。为达到上述目的,本发明提供如下技术方案:利用二维液相分离制备抗体类产品相关杂质的方法,所述抗体类产品是贝伐珠单抗,包括以下步骤:(1)第一维液相色谱进行分离纯化:色谱条件为:采用阳离子交换色谱柱,以ph值为7.0-8.0的缓冲溶液为流动相a,以添加有洗脱剂的ph值为7.0-8.0的缓冲溶液为流动相b,柱温为30-50℃,流速为0.8-1.2ml/min,检测波长为280nm,梯度洗脱条件为:0~5min,4%b;5~24min,4%~12.2%b;24~24.1min,12.2%~100%b;24.1~29min,100%b;29~29.1min,100%~4%b;29.1~37min,4%b;(2)第二维液相色谱进行分离制备:将第一维液相色谱按照保留时间切割所得的馏分引流到第二维液相色谱,进行分离制备:色谱条件为:采用亲和色谱柱,以添加有洗脱剂的ph值为6.3-7.3的缓冲溶液为流动相a,以ph值为2.5-3.5的缓冲溶液为流动相b,柱温为20-30℃,流速为0.3-0.5ml/min,检测波长为280nm,梯度洗脱条件为:0~3min,0%b;3~3.1min,0%~100%b;3.1~6min,100%b;6~6.1min,100%~0%b;6.1~10min,0%b。进一步地,步骤(2)中,亲和色谱柱填充粒径为1.5-12μm。利于提高第二维液相色谱分离制备的效果。进一步地,步骤(2)中,亲和色谱柱条件为:2.1×50mm。使第二维液相色谱分离制备的效果较好。进一步地,步骤(2)中,洗脱剂为hcl或有机酸。进一步地,步骤(2)中,洗脱剂的浓度为5-150mmol/l。使第二维液相色谱分离制备过程中,各目标峰分离效果较好,从而利于回收率的提高。进一步地,步骤(2)中,流动相a的缓冲溶液为ph值为6.8、浓度为50mmol/l的tris-hcl缓冲溶液。使第二维液相色谱分离制备过程中,各目标峰峰形较佳,从而利于回收率的提高。进一步地,步骤(2)中,流动相b的缓冲溶液为ph值为3.0的缓冲溶液,其由0.1mol/l的甘氨酸加入hcl制得。利于提高各目标峰的分离效果,进而利于回收率的提高。进一步地,步骤(1)中,阳离子交换色谱柱条件为:4.0×250mm。进一步地,步骤(1)中,缓冲溶液为ph值为7.5、浓度为25mmol/l的tris-hcl缓冲溶液。进一步地,步骤(1)中,洗脱剂为450mmol/l的nacl。本发明的有益效果在于:利用二维液相分离制备抗体类产品相关杂质的方法,使杂质样品的回收率得到了显著的提高,从而也节省了大量的样品,尤其对于非常珍贵的样品,其次,本方法大大提高了分离制备相关杂质样品的工作效率,使工作效率提高了10倍以上,节省了大量人力,另外,由于避免了冗长的杂质样品处理过程,避免了杂质样品的二次降解与被修饰,从而提高了分离制备的杂质的纯度。上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合附图详细说明如后。附图说明图1为实施例三中二维液相分离制备贝伐珠单抗相关杂质的第一维液相色谱图;图2为实施例三中分离制备所得物质a1的液相色谱图与图1的对照谱图;图3为实施例三中分离制备所得物质a2的液相色谱图与图1的对照谱图;图4为实施例三中分离制备所得物质a3的液相色谱图与图1的对照谱图;图5为实施例三中分离制备所得物质a4的液相色谱图与图1的对照谱图;图6为实施例三中分离制备所得物质a5的液相色谱图与图1的对照谱图;图7为实施例三中分离制备所得物质主峰mp的液相色谱图与图1的对照谱图;图8为实施例三中分离制备所得物质b1的液相色谱图与图1的对照谱图;图9为实施例三中分离制备所得物质b2的液相色谱图与图1的对照谱图;图10为实施例三中分离制备所得物质a1的质谱图谱;图11为实施例三中分离制备所得物质a2的质谱图谱;图12为实施例三中分离制备所得物质a3的质谱图谱;图13为实施例三中分离制备所得物质a4的质谱图谱;图14为实施例三中分离制备所得物质a5的质谱图谱;图15为实施例三中分离制备所得物质主峰mp的质谱图谱;图16为实施例三中分离制备所得物质b1的质谱图谱;图17为实施例三中分离制备所得物质b2的质谱图谱。具体实施方式下面结合附图和实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但不用来限制本发明的范围。实施例一将贝伐珠单抗样品稀释至20mg/ml制得待测样品,用二维液相分离制备贝伐珠单抗相关杂质,第一维液相色谱进行分离纯化的条件为:采用阳离子交换色谱柱,以ph值为7.0的缓冲溶液为流动相a,以添加有450mmol/l的nacl洗脱剂的ph值为7.0的缓冲溶液为流动相b,柱温为30℃,流速为1ml/min,检测波长为280nm,梯度洗脱条件为:0~5min,4%b;5~24min,4%~12.2%b;24~24.1min,12.2%~100%b;24.1~29min,100%b;29~29.1min,100%~4%b;29.1~37min,4%b;第二维液相色谱进行分离制备的条件为:采用亲和色谱柱,以添加有5mmol/lhcl洗脱剂的ph值为6.3的缓冲溶液为流动相a,以ph值为2.5的缓冲溶液为流动相b,柱温为20℃,流速为0.5ml/min,检测波长为280nm,梯度洗脱条件为:0~3min,0%b;3~3.1min,0%~100%b;3.1~6min,100%b;6~6.1min,100%~0%b;6.1~10min,0%b。由第一维液相色谱的进样阀进样,进样量为100μg,将第一维液相色谱中各目标物质的馏分,通过切换阀,分别引流到第二维液相色谱的八根色谱柱中,如此重复三次,待贝伐珠单抗相关杂质在第二维液相色谱柱累积一定量之后,按照第二维液相色谱进行分离制备的条件将各杂质制备。亲和色谱柱条件为:2.1×50mm,粒径12μm。第二维液相色谱分离制备的条件中,流动相a的缓冲溶液为tris-hcl缓冲溶液,流动相b的缓冲溶液为0.1mol/l的甘氨酸加入hcl制得。阳离子交换色谱柱条件为:4.0×250mm。第一维液相色谱分离制备的条件中,缓冲溶液为tris-hcl缓冲溶液。实验结果表明:制备所得的八种杂质均具有较好的回收率,且纯度较高。实施例二将贝伐珠单抗样品稀释至20mg/ml制得待测样品,用二维液相分离制备贝伐珠单抗相关杂质,第一维液相色谱进行分离纯化的条件为:采用阳离子交换色谱柱,以ph值为8.0的缓冲溶液为流动相a,以添加有450mmol/l的nacl洗脱剂的ph值为8.0的缓冲溶液为流动相b,柱温为50℃,流速为1ml/min,检测波长为280nm,梯度洗脱条件为:0~5min,4%b;5~24min,4%~12.2%b;24~24.1min,12.2%~100%b;24.1~29min,100%b;29~29.1min,100%~4%b;29.1~37min,4%b;第二维液相色谱进行分离制备的条件为:采用亲和色谱柱,以添加有80mmol/lhcl洗脱剂的ph值为7.3的缓冲溶液为流动相a,以ph值为3.5的缓冲溶液为流动相b,柱温为30℃,流速为0.3ml/min,检测波长为280nm,梯度洗脱条件为:0~3min,0%b;3~3.1min,0%~100%b;3.1~6min,100%b;6~6.1min,100%~0%b;6.1~10min,0%b。由第一维液相色谱的进样阀进样,进样量为100μg,将第一维液相色谱中各目标物质的馏分,通过切换阀,分别引流到第二维液相色谱的八根色谱柱中,如此重复五次,待贝伐珠单抗相关杂质在第二维液相色谱柱累积一定量之后,按照第二维液相色谱进行分离制备的条件将各杂质制备。亲和色谱柱条件为:2.1×50mm,填充粒径1.5μm。第二维液相色谱分离制备的条件中,流动相a的缓冲溶液为tris-hcl缓冲溶液,流动相b的缓冲溶液为0.1mol/l的甘氨酸加入hcl制得。阳离子交换色谱柱条件为:4.0×250mm。第一维液相色谱分离制备的条件中,缓冲溶液为tris-hcl缓冲溶液。实验结果表明:制备所得的八种杂质均具有较好的回收率,且纯度较高。实施例三将贝伐珠单抗样品稀释至20mg/ml制得待测样品,用二维液相分离制备贝伐珠单抗相关杂质,第一维液相色谱进行分离纯化的条件为:采用阳离子交换色谱柱,以ph值为7.5的缓冲溶液为流动相a,以添加有450mmol/l的nacl洗脱剂的ph值为7.5的缓冲溶液为流动相b,柱温为40℃,流速为1ml/min,检测波长为280nm,梯度洗脱条件为:0~5min,4%b;5~24min,4%~12.2%b;24~24.1min,12.2%~100%b;24.1~29min,100%b;29~29.1min,100%~4%b;29.1~37min,4%b;第二维液相色谱进行分离制备的条件为:采用亲和色谱柱,以添加有150mmol/l乙酸洗脱剂的ph值为6.8的缓冲溶液为流动相a,以ph值为3.0的缓冲溶液为流动相b,柱温为25℃,流速为0.5ml/min,检测波长为280nm,梯度洗脱条件为:0~3min,0%b;3~3.1min,0%~100%b;3.1~6min,100%b;6~6.1min,100%~0%b;6.1~10min,0%b。亲和色谱柱条件为:2.1×50mm,粒径5μm。第二维液相色谱分离制备的条件中,流动相a的缓冲溶液为50mmol/l的tris-hcl缓冲溶液,流动相b的缓冲溶液为0.1mol/l的甘氨酸加入hcl制得。阳离子交换色谱柱条件为:4.0×250mm。第一维液相色谱分离制备的条件中,缓冲溶液为25mmol/l的tris-hcl缓冲溶液。由第一维液相色谱的进样阀进样,进样量为100μg,将第一维液相色谱中各目标物质的馏分,通过切换阀,分别引流到第二维液相色谱的八根色谱柱中,第一维液相色谱中八种目标物质的馏分引流到第二维液相色谱的时间以相对保留时计分别为:物质a10.65-0.74;物质a20.79-0.81;物质a30.87-0.88;物质a40.90-0.93;物质a50.94-0.96;物质mp0.98-1.02;物质b11.05-1.09;物质b21.10-1.13,然后,按照第二维液相色谱进行制备的条件将各杂质制备,分离制备得a1、a2、a3、a4、a5、主峰mp、b1和b2八种物质,第一维液相色谱图见图1。将分离制备得到的八种物质依照第一维液相色谱的条件进行分析,并与原第一维液相色谱图进行对照,所得对照谱图见图2-图9,结果显示,制备所得的a1、a2、a3、a4、a5、主峰mp、b1和b2八种物质的保留时间与原第一维液相色谱图中各物质的保留时间一致,且八种物质均为单一色谱峰,制备所得各物质回收率和纯度如下述表一所示。表一杂质种类制备体积(μl)回收率(%)纯度(%)a1170-22082.899.04a2170-22075.993.08a3170-22078.695.50a4170-22068.897.04a5170-22065.297.15主峰mp170-22085.9100.00b1170-22076.594.52b2170-22077.691.16从上述表一可以看出,制备所得的八种杂质均具有较好的回收率,且纯度较高,均达到了90%以上。对制备所得的a1、a2、a3、a4、a5、主峰mp、b1和b2八种物质进行质谱分析,所得质谱图谱见图10-图17。以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1