一种砂仁药材指纹图谱的检测方法与流程
本发明涉及砂仁药材检测分析
技术领域:
,具体涉及一种砂仁药材指纹图谱的检测方法。
背景技术:
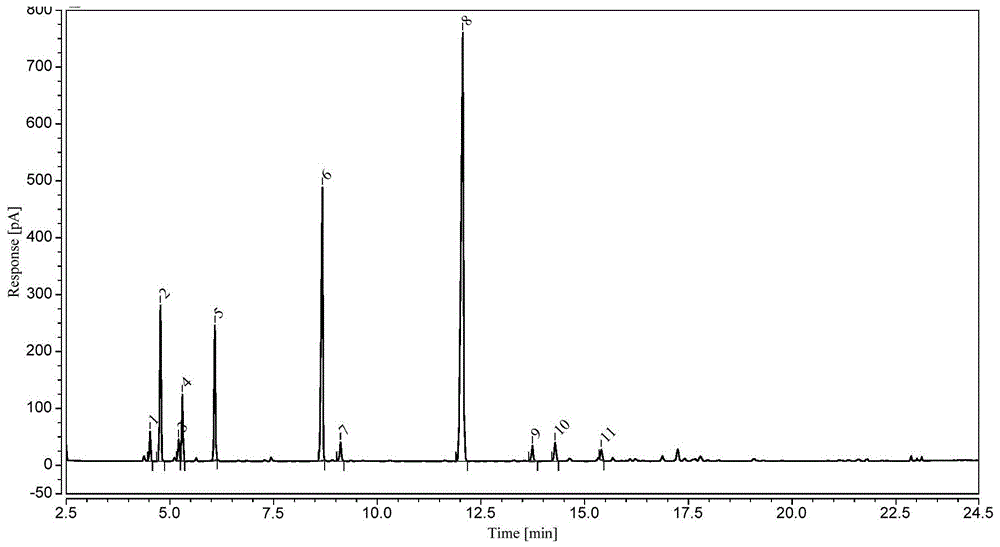
:砂仁为我国著名的“四大南药”之一,可以食用也可以入药,因具有化湿开胃、温脾止泻、理气安胎的功效,在中医临床上具有广泛的应用价值。但其成分的种类及含量易受到产地、品种、采收等众多因素的影响。正是由于砂仁对生态环境的特殊选择以及授粉困难等因素,产量较低,商品供不应求,价格高涨,导致市场上出现不同程度的伪品,常以在外观性状上相似的豆蔻、草果、草豆蔻等近缘植物的种子或果实混充砂仁。在砂仁品种上,目前广东阳春产砂仁质量最佳,素有道地药材之称,绿壳砂主产于越南、缅甸,海南砂主产于海南,这三种砂仁被药典列为是正品砂仁。但在药典中,含量测定仅以测定乙酸龙脑酯一种成分来控制砂仁的质量状态,不能完整反应砂仁药材的全貌,因此需要可以提供丰富化合物信息的检测方法全面表征砂仁药材的质量。目前针对砂仁真伪及质量控制主要是从性状鉴定、显微鉴定、薄层色谱鉴别、挥发油含量鉴定、高效液相色谱指纹图谱鉴别等方式,单纯采用其中某种方式存在鉴别不够准确或操作繁琐、需要精密仪器的缺陷。因此,亟需更加简单、有效、合理的质量评价方法以确保砂仁使用的安全性和有效性。中药指纹图谱是运用一定综合性技术的分析方法,通过特征性色谱图或光谱图,在整体上对其化学特征进行表征,是被公认为最有效的综合性评价中药的整体质量的技术手段之一,目前关于砂仁药材指纹图谱检测的相关专利并不多。技术实现要素:本发明目的在于提供一种砂仁药材指纹图谱的检测方法,该方法能够简便、快速获得砂仁药材的指纹图谱,实现对砂仁药材中包括α-蒎烯、莰烯、β-月桂烯、d-柠檬烯、樟脑、龙脑、乙酸龙脑酯、丁香酚、石竹烯在内的共11个特征峰进行快速准确的定性,通过将砂仁药材供试品与对照品指纹图谱进行对比计算相似度,可以快速有效评价样品的质量,并且具有基线平稳、噪音干扰少、检测重复性好、色谱峰信息多、灵敏度高的特点。本发明的技术方案如下:一种砂仁药材指纹图谱的检测方法,包括下述步骤:1)将砂仁药材冷藏保存后粉碎过筛,与提取溶剂按照1~5g:10~30ml的比例混合,形成样液;2)将上述步骤1)得到的样液超声提取10~45min,与净化剂按照1ml:4~50mg的比例混合;所述净化剂为psa和石墨化碳的混合物,所述psa和石墨化碳的质量比为1:1~5;3)将所述步骤2)得到的上清液用微孔滤膜过滤,得供试品溶液;4)将所述步骤3)得到的供试品溶液进行气相色谱分析,获得色谱图和特征峰的保留时间;5)将c8-c20正构烷烃标准溶液采用与步骤4)同样的色谱条件,进行气相色谱分析,获得色谱图和c8-c20正构烷烃的保留时间;6)将所述步骤4)中特征峰的保留时间和步骤5)中c8-c20正构烷烃的保留时间代入公式,计算特征峰的保留指数;7)将所述步骤6)所得的保留指数与理论保留指数进行比对,对特征峰进行定性;8)将所述步骤7)中定性后的特征峰化合物用乙酸乙酯溶解,制备成对照品混合溶液;9)将所述步骤8)得到的对照品混合溶液采用与步骤4)同样的色谱条件,进行气相色谱分析,获得对照品色谱图和保留时间;10)将所述步骤9)获得的对照品色谱图和保留时间,对步骤4)的特征峰进行定性确认;11)将10批不同批次的同品种砂仁药材按步骤1)~步骤4)进行分析,对特征峰进行指认,并将色谱图导入中药指纹图谱数据库进行相似度评价,生成对照指纹图谱;12)将待测砂仁药材样品按步骤1)~步骤4)进行分析,对特征峰进行指认,并将色谱图导入中药指纹图谱数据库进行相似度评价,与所述步骤11)中得到的对照指纹图谱进行对比,计算相似度。优选的,所述步骤1)中提取溶剂为乙酸乙酯,砂仁药材粉末与乙酸乙酯按照5g:25ml的比例混合。优选的,所述步骤2)中超声提取30min,提取溶剂乙酸乙酯的体积与净化剂的质量比为25ml:550mg;psa和石墨化碳的质量比为1:1.75。优选的,所述步骤4)中气相色谱分析参数包括色谱柱固定相为(5%-苯基)-甲基聚硅氧烷;进样口温度为250℃,载气:氮气,纯度≥99.999%,分流比为10:1,恒流模式,流速1.0ml/min,进样量1μl;柱温初温为80℃,保持1min,以5℃/min升至180℃,再以30℃/min升至270℃,保持9min;检测器:氢火焰离子化检测器,温度为260℃。优选的,所述步骤4)中指纹图谱共有11个特征峰,所述特征峰与参照峰8号峰的相对保留时间为:1号峰:0.3752±0.0003;2号峰:0.3960±0.0004;3号峰:0.4324±0.0004;4号峰:0.4399±0.0003;5号峰:0.5048±0.0003;6号峰:0.7195±0.0002;7号峰:0.7558±0.0004;8号峰为参照峰(p8):1.0000;9号峰:1.1389±0.0001;10号峰:1.1845±0.0003;11号峰:1.2766±0.0003。优选的,保留指数计算公式为ri=100×{n+[t(x)-t(n)]/[t(n+1)-t(n)]},式中ri为保留指数;n为正构烷烃的碳原子数;t(n+1)、t(n)分别代表碳数为n+1、n的正构烷烃的保留时间(min);t(x)为被测组分保留时间(min),且t(n+1)>t(x)>t(n)。经保留指数计算与比对,所述步骤4)供试品溶液中1-11号峰分别为1号峰:α-蒎烯;2号峰:莰烯;3号峰:β-蒎烯;4号峰:β-月桂烯;5号峰:d-柠檬烯,6号峰:樟脑;7号峰:龙脑;8号峰:乙酸龙脑酯;9号峰:丁香酚;10号峰:α-古巴烯;11号峰:石竹烯。相对于现有技术,本发明具有以下有益效果:(1)将砂仁药材粉末采用乙酸乙酯进行提取,对药材成分提取效果较好且溶剂毒性较小;(2)采用乙二胺-n-丙基硅烷、石墨化碳进行净化,在不影响特征峰响应的前提下,大大减少了样品对衬管等仪器部件的污染风险,从而降低了仪器运行成本;(3)采用正构烷烃保留指数法进行特征峰定性,具有指认峰数量多、方便快捷、成本低的优势;(4)本发明具有操作简便、检测限低、结果可靠的优点,可以快速、准确鉴别砂仁药材质量优劣、真伪、稳定性及道地性。附图说明图1为实施例1中供试品溶液的色谱图;图2为实施例1中c8-c20正构烷烃标准溶液的色谱图;图3为实施例1中对照品混合溶液的色谱图;图4为实施例2中乙醇提取供试品溶液色谱图;图5为实施例2中乙酸乙酯提取供试品溶液色谱图;图6为实施例3中净化条件下供试品溶液的色谱图;图7为实施例3中未净化条件下供试品溶液的色谱图;图8为实施例4中专属性试验中空白溶剂对照溶液的色谱图;图9为实施例5中10批砂仁药材的色谱图;图10为实施例5中10批砂仁药材的对照指纹图谱;图11为实施例6中样品(s11、s12)与对照指纹图谱的对比图。具体实施方式下面结合实施例对本发明提供的砂仁药材指纹图谱检测方法及指纹图谱与应用进行详细说明,但是不能把它们理解为对本发明保护范围的限定。凡基于本发明上述内容所实现的技术均属于本发明的范围。实施例1色谱图特征峰的指认供试品溶液制备:称取5g砂仁药材粉末于50ml离心管中,加入25ml乙酸乙酯,超声提取30min,加入200mg乙二胺-n-丙基硅烷、350mg石墨化碳进行净化,上清液用0.22μm微孔滤膜过滤,即得供试品溶液。c8-c20正构烷烃标准溶液:购于上海安谱实验科技股份有限公司。将供试品溶液与c8-c20正构烷烃标准溶液采用相同的色谱条件注入气相色谱仪,供试品溶液色谱图分别见图1,c8-c20正构烷烃色谱图见图2。采用公式计算特征峰的保留指数,并与文献报道的保留指数进行比对,对特征峰进行定性,结果见表1。保留指数计算公式为ri=100×{n+[t(x)-t(n)]/[t(n+1)-t(n)]},其中,ri为保留指数;n为正构烷烃的碳原子数;t(n+1)、t(n)分别代表碳数为n+1、n的正构烷烃的保留时间(min);t(x)为被测组分保留时间(min),且t(n+1)>t(x)>t(n)。表1砂仁药材中特征峰保留指数与定性结果结果表明:保留指数测定值与文献参考值差值≤10,鉴定结果可靠。称取表1中定性的化合物标准品,用乙酸乙酯配制成对照品混合溶液,其中α-蒎烯、莰烯、β-月桂烯、d-柠檬烯、樟脑、龙脑、乙酸龙脑酯、丁香酚、石竹烯的浓度分别为79.40mg/l、6.12mg/l、17.21mg/l、19.02mg/l、32.49mg/l、15.22mg/l、175.88mg/l、498.95mg/l、36.22mg/l。将对照品混合溶液采用与供试品溶液相同的色谱条件注入气相色谱仪,得到对照品混合溶液色谱图见图3,根据相对保留时间进一步进行定性确认,结果与保留指数法吻合。实施例2供试品溶液制备中提取溶剂优选实验称取5g砂仁药材粉末于50ml离心管中,共2份,分别加入25ml乙醇和乙酸乙酯,超声提取30min,加入200mg乙二胺-n-丙基硅烷、350mg石墨化碳净化,上清液用0.22μm微孔滤膜过滤,即得供试品溶液,供气相色谱分析。乙醇提取供试品色谱图见图4,乙酸乙酯提取供试品色谱图见图5。根据图4~5的试验结果,采用乙酸乙酯为提取溶剂时,色谱峰信号强度大,提取效果优于乙醇,故优选乙酸乙酯为提取溶剂。实施例3供试品溶液制备中净化条件优选实验称取5g砂仁药材粉末于50ml离心管中,共2份,加入25ml乙酸乙酯,超声30min,其中一份加入200mg乙二胺-n-丙基硅烷、350mg石墨化碳净化,另一份不加,上清液均用0.22μm微孔滤膜过滤供气相色谱分析。净化处理的供试品色谱图见图6,未净化处理的供试品色谱图见图7。根据图6~7的试验结果,净化与不净化对色谱峰的响应无明显影响,但是经过净化的样品,对仪器的污染减少,降低了仪器部件更换频次、仪器故障风险及检测成本,因此优选净化处理过程建立砂仁药材的指纹图谱。实施例4方法学考察1.试剂乙酸乙酯,色谱纯,购于国药集团化学试剂有限公司;石墨化碳(gcb)购自博纳艾杰尔科技;乙二胺-n-丙基硅烷(psa)购自上海安谱实验科技股份有限公司。2.对照品α-蒎烯、莰烯、d-柠檬烯、樟脑、龙脑、乙酸龙脑酯、丁香酚对照品:购于中国食品药品检定研究院;β-月桂烯、石竹烯对照品:购于上海安谱实验科技股份有限公司。3.对照品混合溶液制备分别称取α-蒎烯、莰烯、β-月桂烯、d-柠檬烯、樟脑、龙脑、乙酸龙脑酯、丁香酚、石竹烯对照品适量,加乙酸乙酯溶解,得相应的对照品溶液储备液;再移取适量对照品溶液储备液,加乙酸乙酯溶解,得α-蒎烯、莰烯、β-月桂烯、d-柠檬烯、樟脑、龙脑、乙酸龙脑酯、丁香酚、石竹烯浓度分别为79.40mg/l、6.12mg/l、17.21mg/l、19.02mg/l、32.49mg/l、15.22mg/l、175.88mg/l、498.95mg/l、36.22mg/l的对照品混合溶液。4.供试品溶液制备将冷藏保存后的砂仁样品粉碎过筛,称取5g粉末于50ml离心管中,加入25ml乙酸乙酯,超声30min,加入200mg乙二胺-n-丙基硅烷、350mg石墨化碳净化,上清液用0.22μm微孔滤膜过滤,供gc检测。5.色谱条件气相色谱参数为色谱柱固定相为(5%-苯基)-甲基聚硅氧烷;进样口温度为250℃,载气:氮气,纯度≥99.999%,分流比为10:1,恒流模式,流速1.0ml/min,进样量1μl;柱温初温为80℃,保持1min,以5℃/min升至180℃,再以30℃/min升至270℃,保持9min;检测器:氢火焰离子化检测器,温度为260℃;氢气流速35ml/min,空气流速350ml/min,尾吹氮气流速30ml/min。在优选的条件下,注入对照品混合溶液。结果表明,乙酸龙脑酯色谱峰峰面积较大,且是砂仁成分的典型指标,因此选择乙酸龙脑酯峰作为参照峰。系统适应性试验:在上述色谱条件下,取对照品混合溶液重复进样6次,记录相对保留时间(rrt)与相对峰面积(rpa),并计算rsd(%),结果见表2、表3。表2系统适应性试验结果(相对保留时间)编号p1p2p4p5p6p7p8p9p1110.37770.39850.44250.50750.72110.76041.00001.14551.283720.37730.39820.44230.50710.72090.76041.00001.14561.284030.37780.39840.44250.50740.72120.76041.00001.14551.283840.37720.39800.44200.50700.72090.76021.00001.14541.283650.37730.39810.44230.50720.72100.76031.00001.14551.283860.37710.39790.44210.50710.72100.76021.00001.14551.2838均值0.37740.39820.44230.50720.72110.76031.00001.14551.2838rsd(%)0.07040.05800.04360.03860.01510.01100.00000.00790.0109表3系统适应性试验结果(相对峰面积)编号p1p2p4p5p6p7p8p9p1110.16370.15480.20100.16720.48970.11981.00000.59320.095920.16380.15270.20080.16790.48870.12111.00000.59850.096230.16250.15190.20200.16750.48880.12101.00000.59640.096340.16480.15330.20240.16780.48850.12211.00000.59920.096950.16190.15400.20060.16660.48800.12131.00000.59890.095960.16480.15560.20390.16900.49020.12161.00000.59910.0959均值0.16360.15370.20180.16770.48900.12121.00000.59760.0962rsd(%)0.73260.89120.62040.47410.16930.62850.00000.39460.4139结果表明:对照品混合溶液的重复性rsd小于5%,系统适应性良好。专属性试验:在上述色谱图条件下,取上述对照品混合溶液、供试品溶液、空白溶剂对照溶液各进一针,记录色谱图,空白溶剂对照溶液色谱图见图8。结果表明:空白溶剂对照无干扰,方法专属性良好。精密度试验:在上述色谱条件下,取供试品溶液连续进样6次,以乙酸龙脑酯色谱峰(p8)为对照,计算11个特征峰的相对保留时间rsd(%)与相对峰面积rsd(%),结果见表4和表5。表4精密度试验结果(相对保留时间)特征峰123456均值rsd(%)p10.37510.37530.37510.37510.37560.37530.37530.0472p20.39600.39600.39610.39600.39630.39630.39610.0403p30.43240.43240.43260.43250.43270.43280.43260.0356p40.43990.44000.43990.43990.44030.44020.44000.0426p50.50480.50480.50490.50470.50500.50500.50490.0267p60.71930.71950.71950.71950.71970.71980.71950.0230p70.75580.75590.75580.75590.75610.75620.75590.0240p81.00001.00001.00001.00001.00001.00001.00000.0000p91.13921.13891.13891.13941.13871.13891.13900.0208p101.18451.18461.18451.18471.18431.18461.18450.0120p111.27671.27681.27631.27691.27651.27681.27670.0194表5精密度试验结果(相对峰面积)结果表明:色谱图11个共有特征峰的相对保留时间、相对峰面积的rsd均小于5%,符合指纹图谱制订的相关要求。稳定性试验:取同一批样品,按上述方法制备供试品溶液,室温下放置0、2、4、8、12、24h进样,以乙酸龙脑酯色谱峰(p8)为参照,计算各特征峰的相对保留时间和相对峰面积rsd(%),结果见表6和表7。表6稳定性试验结果(相对保留时间)特征峰02481224均值rsd(%)p10.37500.37510.37510.37560.37580.37540.37530.0847p20.39570.39590.39610.39650.39650.39620.39620.0746p30.43210.43220.43240.43290.43290.43260.43250.0806p40.43970.43980.43980.44040.44040.44020.44010.0687p50.50450.50460.50470.50520.50510.50500.50490.0599p60.71920.71930.71930.71990.71960.71970.71950.0385p70.75560.75560.75570.75620.75610.75600.75590.0345p81.00001.00001.00001.00001.00001.00001.00000.0000p91.13891.13881.13871.13891.13891.13861.13880.0112p101.18421.18471.18421.18441.18431.18411.18430.0186p111.27651.27681.27631.27641.27651.27601.27640.0210表7稳定性试验结果(相对峰面积)试验结果表明:色谱图各共有特征峰的相对保留时间、相对峰面积rsd均小于5%,符合指纹图谱制订的相关要求。重复性试验:取同一批样品,按上述方法制备6份供试品溶液,依次进样,以乙酸龙脑酯色谱峰(p8)为参照,计算各共有特征峰的相对保留时间和相对峰面积rsd(%),结果见表8和表9。表8重复性试验结果(相对保留时间)特征峰123456均值rsd(%)p10.37490.37490.37520.37520.37550.37540.37520.0672p20.39560.39580.39610.39610.39650.39630.39600.0795p30.43210.43210.43250.43250.43280.43270.43240.0678p40.43970.43970.44000.44000.44020.44020.43990.0513p50.50450.50450.50470.50480.50500.50500.50480.0465p60.71940.71920.71950.71960.71950.71970.71950.0260p70.75540.75560.75580.75580.75610.75600.75580.0309p81.00001.00001.00001.00001.00001.00001.00000.0000p91.13901.13881.13891.13871.13901.13871.13890.0112p101.18461.18471.18461.18421.18461.18411.18450.0208p111.27691.27701.27651.27651.27651.27651.27660.0181表9重复性试验结果(相对峰面积)试验结果表明:色谱图各共有特征峰的相对保留时间、相对峰面积rsd均小于5%,符合指纹图谱制订的相关要求。实施例5砂仁药材对照指纹图谱的建立将10批代表性砂仁药材(编号为s1~s10)按实施例4的方法制备成供试品溶液,注入气相色谱分析,获得10批样品色谱图,见图9,导入中药色谱指纹图谱相似度评价系统进行数据比对处理,生成对照指纹图谱,见图10。表10砂仁药材相似度计算结果编号s1s2s3s4s5s6s7s8s9s10对照s11.0001.0000.9940.9920.9940.9980.9980.9950.9970.9970.998s21.0001.0000.9940.9920.9940.9980.9980.9950.9970.9970.998s30.9940.9941.0000.9990.9990.9970.9970.9980.9950.9950.998s40.9920.9920.9991.0001.0000.9960.9960.9980.9940.9940.998s50.9940.9940.9991.0001.0000.9980.9970.9990.9950.9950.999s60.9980.9980.9970.9960.9981.0001.0000.9990.9990.9991.000s70.9980.9980.9970.9960.9971.0001.0000.9990.9990.9991.000s80.9950.9950.9980.9980.9990.9990.9991.0000.9990.9990.999s90.9970.9970.9950.9940.9950.9990.9990.9991.0001.0000.999s100.9970.9970.9950.9940.9950.9990.9990.9991.0001.0000.999对照0.9980.9980.9980.9980.9991.0001.0000.9990.9990.9991.000选择11个共有峰作为指纹图谱的特征峰,以乙酸龙脑酯色谱峰(p8)为参照,计算相似度,结果见表10。结果显示,10批代表性砂仁样品间的相似度均大于0.99,表明这10批砂仁样品质量一致,符合制订对照指纹图谱的要求。实施例6指纹图谱在砂仁药材质量控制中的应用将2批砂仁药材样品(s11、s12)按实施例4的方法制备供试品溶液,注入气相色谱分析,获得2批样品色谱图,导入中药色谱指纹图谱相似度评价系统(由国家药典委员会发布),同时导入实施例5对照指纹图谱,选择11个共有峰作为指纹图谱的特征峰,以乙酸龙脑酯色谱峰(p8)为参照(s),计算s11、s12与对照指纹图谱的相似度,相似度结果见表11。表112批砂仁药材相似度计算结果编号s11s12对照s111.0001.0000.998s121.0001.0000.998对照0.9980.9981.000结果显示,药材s11、s12的相似度均为0.998,表明该两批砂仁药材质量合格稳定。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1