具有改善的储存稳定性的药物组合物及其制备方法与流程
本发明涉及具有改善的储存稳定性的药物组合物及其制备方法,更具体地涉及包含两亲性嵌段共聚物的水溶性差的药物的药物组合物,其中特定相关化合物的含量保持在规定限量以内;并且涉及其制备方法。
背景技术:
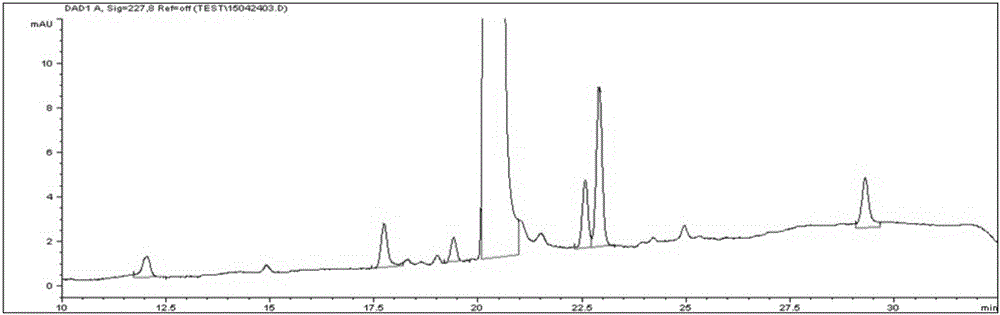
:水溶性差的药物的溶解是经由口服或肠胃外给药将药物递送到体内的关键技术。这种溶解方法包括在水溶液中添加表面活性剂以形成胶束,然后在其中包埋(entrapping)水溶性差的药物的方法。用作表面活性剂的两亲性嵌段共聚物包含亲水性聚合物嵌段和疏水性聚合物嵌段。由于亲水性聚合物嵌段在体内直接接触血液蛋白质和细胞膜,因此已使用具有生物相容性的聚乙二醇或单甲氧基聚乙二醇等。疏水性聚合物嵌段改善与疏水性药物的亲和性,因此已使用具有可生物降解的聚丙交酯、聚乙交酯、聚(乳酸-乙交酯)(poly(lactic-glycolide))、聚己内酯、聚氨基酸或聚原酸酯等。特别地,因为聚丙交酯衍生物具有优异的生物相容性并且在体内水解为无害的乳酸,因此它们已经以各种形式应用于药物载体。聚丙交酯衍生物随其分子量变化具有不同的物理性质,并且已经开发出各种形式,例如微球、纳米颗粒、聚合物凝胶和植入剂。第6,322,805号美国专利公开了用于递送水溶性差的药物的组合物,其由聚合物胶束型药物载体和水溶性差的药物组成,其中所述聚合物胶束型药物载体由二嵌段共聚物或三嵌段共聚物形成,所述二嵌段共聚物或三嵌段共聚物不由交联剂交联,其由选自聚丙交酯、聚乙交酯、聚(丙交酯-乙交酯)、聚己内酯及其衍生物的至少一种可生物降解的疏水性聚合物和聚(环氧烷)作为亲水性聚合物组成,其中将水溶性差的药物物理地包埋在药物载体中并溶解,并且其中所述聚合物胶束型药物载体在水中形成透明水溶液并将水溶性差的药物有效地递送到体内。根据上述美国专利,聚乙二醇-聚丙交酯二嵌段共聚物是通过以下步骤合成的:从单甲氧基聚乙二醇中除去水分,向其中加入溶解在甲苯中的辛酸亚锡并减压除去甲苯,向所得混合物中加入D,L-丙交酯并进行聚合反应,加入氯仿以溶解所产生的嵌段共聚物,在搅拌下小份地滴加过量的乙醚以形成沉淀,过滤形成的沉淀,并用乙醚洗涤数次。然而,该方法难以在大规模生产中使用,因此不可商用。此外,用于纯化的醚可能残留在最终的聚合物胶束组合物中。第8,853,351号美国专利公开了一种制备两亲性嵌段共聚物的方法,其包括(a)将两亲性嵌段共聚物溶解在水混溶性有机溶剂中;(b)向步骤(a)中获得的聚合物溶液中加入并混合碱金属盐(碳酸氢钠、碳酸钠、碳酸氢钾、碳酸钾或碳酸锂)的水溶液;(c)通过盐析对步骤(b)中获得的溶液进行有机相和水相分离;和(d)分离步骤(c)中获得的有机相并除去其中的有机溶剂以回收聚合物。然而,该方法涉及复杂的步骤,需要额外的步骤以除去碱金属盐和用于盐析的盐(氯化钠或氯化钾),并且即使在将其除去后也可能具有残留的金属盐。必须在各个方面严格控制药物的杂质。特别地,在杂质源于活性药物成分(API)的情况下,每个国家在其药物审批指南中设定了药物产品中源自API的、已知或未知杂质(相关化合物)的量的上限。此外,还有一些国际上使用的标准,ICH指南Q3A是有代表性的一个。在该指南中,在审批药物时,药物中每种相关化合物的量限制为最多0.1%或0.2%等,并且根据超过限量的相关化合物,有差别地适用应当规定的诸如毒性相关数据等的信息。这意味着在制备药物的过程中必须减少相关化合物的量,因为药物的相关化合物在体内如何作用是未知的。因此,用于减少相关化合物的制造方法和根据每种相关化合物的特性(结构和毒性)设定其量的上限是药物质量控制中的必要因素。技术实现要素:技术问题本发明的一个目的为提供含有两亲性嵌段共聚物的水溶性差的药物的聚合物胶束型药物组合物,其含有在规定限量以内的量的特定相关化合物。本发明的另一个目的为提供制备所述药物组合物的方法。技术手段本发明的一方面提供了聚合物胶束药物组合物,其包含:包含亲水性嵌段(A)和疏水性嵌段(B)的纯化的两亲性嵌段共聚物,以及一种或多种水溶性差的药物,其选自紫杉醇和多西紫杉醇,其中当所述药物组合物在40℃下储存6个月时,其含有以所述水溶性差的药物的初始量为100重量份计的小于0.17重量份的由下式1表示的相关化合物:[式1]其中:R1为H或COCH3,R2为苯基或O(CH3)3。本发明的另一方面提供了制备聚合物胶束药物组合物的方法,其包括:(a)纯化包含亲水性嵌段(A)和疏水性嵌段(B)的两亲性嵌段共聚物;(b)将选自紫杉醇和多西紫杉醇的一种或多种水溶性差的药物和纯化的两亲性嵌段共聚物溶解在有机溶剂中;和(c)向步骤(b)中获得的溶液中加入水性溶剂以形成聚合物胶束;当所述药物组合物在40℃下储存6个月时,其含有以所述水溶性差的药物的初始量为100重量份计的小于0.17重量份的由上式1表示的相关化合物。有益效果根据本发明,可以获得具有减少的相关化合物和改善的储存稳定性的水溶性差的药物的药物组合物。附图说明图1为对实验例1中使用的、经过六个月加速试验的、含有紫杉醇的聚合物胶束组合物进行HPLC分析所得的色谱图。图2显示实验例1中获得的相关化合物(RRT0.87±0.02(0.85~0.89),其在下文中可与RRT0.87互换使用)的LC/MS/MS分析结果。图3显示对实验例3中用酸处理紫杉醇所得的混合物中在RRT0.87处获得的物质进行LC/MS/MS分析的结果。图4显示对实验例3中用酸处理紫杉醇所得的混合物中在RRT0.87处获得的物质进行LC/MS/MS分析的产物离子扫描的结果,以及经过六个月加速试验的含有紫杉醇的聚合物胶束药物组合物样品的结果:(a)六个月加速试验后聚合物胶束组合物的HPLC色谱图;(b)通过用酸处理紫杉醇在RRT0.87处所得物质的色谱图。图5显示对实验例3中用酸处理紫杉醇所得的混合物中在RRT0.87处获得的物质进行NMR分析的1HNMR分析结果。图6显示对实验例3中用酸处理紫杉醇所得的混合物中在RRT0.87处获得的物质进行NMR分析的13CNMR分析结果。图7显示对实验例3中用酸处理紫杉醇所得的混合物中在RRT0.87处获得的物质进行NMR分析的COSY(相关光谱)分析结果。图8显示对实验例3中用酸处理紫杉醇所得的混合物中在RRT0.87处获得的物质进行NMR分析的HMBC(异核多键相关光谱)分析结果。图9是在实验例5中进行HPLC分析所得的色谱图。具体实施方式下面对本发明进行更详细地说明。本发明实施方案的药物组合物包含含有亲水性嵌段(A)和疏水性嵌段(B)的纯化的两亲性嵌段共聚物。根据本发明的一个实施方案,所述两亲性嵌段共聚物包含由亲水性嵌段(A)和疏水性嵌段(B)组成的A-B型二嵌段共聚物或B-A-B型三嵌段共聚物。根据本发明的一个实施方案,所述两亲性嵌段共聚物可包含,以该共聚物的总重量计,20-95重量%、更具体地40-95重量%的亲水性嵌段。此外,所述两亲性嵌段共聚物可包含,以该共聚物的总重量计,5-80重量%、更具体地5-60重量%的疏水性嵌段。根据本发明的一个实施方案,所述两亲性嵌段共聚物的数均分子量可以为1,000-50,000道尔顿,更具体地为1,500-20,000道尔顿。根据本发明的一个实施方案,所述亲水性嵌段是具有生物相容性的聚合物并且可包含选自聚乙二醇或其衍生物、聚乙烯吡咯烷酮、聚乙烯醇、聚丙烯酰胺及其组合中的一种或多种,更具体地,其可包含选自聚乙二醇、单甲氧基聚乙二醇及其组合中的一种或多种。亲水性嵌段的数均分子量可以为200-20,000道尔顿,更具体地为200-10,000道尔顿。根据本发明的一个实施方案,所述疏水性嵌段是具有生物可降解性的聚合物,并且可以为衍生自α-羟基酸的单体的聚合物。具体地,其可包含选自聚丙交酯、聚乙交酯、聚扁桃酸、聚己内酯、聚二噁烷-2-酮、聚氨基酸、聚原酸酯、聚酐、聚碳酸酯及其组合的一种或多种,更具体地,其可包含选自聚丙交酯、聚乙交酯、聚己内酯、聚二噁烷-2-酮及其组合中的一种或多种。疏水性嵌段的数均分子量可以为200-20,000道尔顿,更具体地为200-10,000道尔顿。根据本发明的一个实施方案,包含聚(α-羟基酸)疏水性聚合物嵌段的两亲性嵌段共聚物可以通过已知的开环聚合方法合成,所述方法使用具有羟基的亲水性聚合物作为引发剂和α-羟基酸内酯单体。例如,以具有羟基的亲水性聚乙二醇或单甲氧基聚乙二醇为引发剂,L-丙交酯或D,L-丙交酯可以通过开环聚合。根据作为引发剂的亲水性嵌段中存在的羟基的数目,可以合成二嵌段共聚物或三嵌段共聚物。在开环聚合中,可以使用有机金属催化剂,例如氧化锡、氧化铅、辛酸锡、辛酸锑等,在制备医疗用聚合物时优选使用具有生物相容性的辛酸锡。在本发明的实施方案中,使用纯化的共聚物作为两亲性嵌段共聚物。根据本发明的优选实施方案,两亲性嵌段共聚物是已通过升华纯化的共聚物。升华提纯可以在优选80℃以上且低于120℃、更优选80-100℃的温度下,以及在优选10托以下、更优选5托以下、更加优选1托以下的真空度的压力下进行优选10-74小时、更优选10-48小时、更加优选24-48小时。在这种条件下通过升华进行纯化可以使共聚物的分子量变化最小化并从共聚物中除去杂质。本发明的药物组合物包含选自紫杉醇和多西紫杉醇的一种或多种水溶性差的药物作为活性成分。根据本发明的一个实施方案,药物组合物还可包含作为另外的活性成分的除紫杉醇和多西紫杉醇之外的一种或多种水溶性差的药物。作为这样的另外的活性成分,可以使用一种或多种选自7-表紫杉醇、t-乙酰基紫杉醇、10-去乙酰基紫杉醇、10-去乙酰基-7-表紫杉醇、7-木糖基紫杉醇、10-去乙酰基-7-戊二酰紫杉醇、7-N,N-二甲基甘氨酰紫杉醇、7-L-丙氨酰紫杉醇和卡巴他赛(cabazitaxel)的紫杉烷抗癌剂。本发明的实施方案的药物组合物可包含,基于100重量份的两亲性嵌段共聚物,0.1-50重量份、更具体地0.5-30重量份的水溶性差的药物。与两亲性嵌段共聚物的量相比,如果水溶性差的药物的量太少,则每种药物使用的两亲性共聚物的重量比较高,因此重构时间会增加。另一方面,如果水溶性差的药物的量太多,则可能存在水溶性差的药物快速沉淀的问题。如本文所用的水溶性差的药物的“初始”量是指制备药物组合物时掺入的水溶性差的药物的重量。在本发明的实施方案中,当所述药物组合物在加速条件下(40℃)储存6个月时,其含有,以所述水溶性差的药物的初始量为100重量份计,小于0.17重量份的由下式1表示的相关化合物:[式1]其中:R1为H或COCH3,R2为苯基或O(CH3)3。根据本发明的一个实施方案,水溶性差的药物为紫杉醇,相关化合物可包括下式1a表示的化合物:[式1a]当在加速条件下(40℃)储存6个月时,本发明实施方案的药物组合物可含有,以所述水溶性差的药物的初始量为100重量份计,小于0.17重量份,优选0.15重量份以下、更优选0.1重量份以下、更加优选0.08重量份以下、最优选0.04重量份以下的式1(特别是式1a)的相关化合物。当在严格条件下(80℃)储存3周时,本发明实施方案的药物组合物可含有,以所述水溶性差的药物的初始量为100重量份计,小于0.76重量份、优选0.5重量份以下、更优选0.4重量份以下、更加优选0.2重量份以下、最优选0.15重量份以下的式1(特别是式1a)的相关化合物。在本发明的实施方案中,含有规定限量以内的量的特定相关化合物的药物组合物是商业可获得的组合物,因为其可以大规模生产。在一个实施方案中,本发明的药物组合物完全不含醚,例如乙醚。在一个实施方案中,本发明的药物组合物完全不具有金属盐,例如碱金属盐和/或用于盐析的盐,例如NaCl或KCl。本发明实施方案的药物组合物可以通过包括以下步骤的方法制备:(a)纯化包含亲水性嵌段(A)和疏水性嵌段(B)的两亲性嵌段共聚物;(b)将选自紫杉醇和多西紫杉醇的一种或多种水溶性差的药物以及纯化的两亲性嵌段共聚物溶解在有机溶剂中;和(c)向步骤(b)中获得的溶液中加入水性溶剂以形成聚合物胶束。两亲性嵌段共聚物的纯化如上所述,可用常规方法形成聚合物胶束。在本发明实施方案的制备药物组合物的方法中,可以使用例如选自醇(例如乙醇)、丙酮、四氢呋喃、乙酸、乙腈和二氧六环及其组合的水混溶性有机溶剂作为有机溶剂,但并不限于此。另外,可以使用选自常规水、蒸馏水、注射用蒸馏水、生理盐水、5%葡萄糖、缓冲液及它们的组合中的一种作为水性溶剂,但不限于此。本发明实施方案的制备药物组合物的方法还可包括在所述步骤(a)之后除去有机溶剂。在实施方案中,所述方法还可包括加入冻干助剂以冻干胶束组合物。可以为冻干组合物添加冻干助剂以维持饼状。在另一个实施方案中,冻干助剂可以是选自糖和糖醇中的一种或多种。糖可以是选自乳糖、麦芽糖、蔗糖或海藻糖中的一种或多种。糖醇可以是选自甘露醇、山梨醇、麦芽糖醇、木糖醇和乳糖醇中的一种或多种。冻干助剂还可用于促进冻干的聚合物胶束组合物在重构时的均匀溶解。基于冻干组合物的总重量,冻干助剂的含量可以为1-90重量%,特别是1-60重量%,更特别是10-60重量%。通过以下实施例对本发明进行更详细地说明。然而,这些实施例仅试图说明本发明,且实施例不以任何方式对本发明的范围进行限制。实施例制备例1:由单甲氧基聚乙二醇和D,L-丙交酯组成的二嵌段共聚物(mPEG-PDLLA)的合成和通过升华方法纯化将150g单甲氧基聚乙二醇(mPEG,数均分子量=2,000)加入到配备有搅拌器的500ml圆底烧瓶中,并在120℃真空条件下搅拌2小时以除去水分。将溶解在200μl甲苯中的0.15g辛酸锡(Sn(Oct)2)加入到反应烧瓶中,并在真空条件下再搅拌1小时以蒸馏并除去甲苯。然后加入150gD,L-丙交酯并在氮气氛下搅拌以溶解。在D,L-丙交酯完全溶解之后,将反应器紧密密封,并在120℃下进行聚合反应10小时。反应终止后,在磁棒搅拌下,将反应器连接到真空泵,产物在1托或更低的压力下通过升华方法纯化7小时,得到262g熔融状态的mPEG-PDLLA。通过用1H-NMR分析获得关于单甲氧基聚乙二醇的末端基团-OCH3的适当峰的相对强度,从而计算分子量(Mn:~3740)。制备例2:通过升华方法纯化二嵌段共聚物(mPEG-PDLLA)将在制备例1的聚合反应过程中获得的进行纯化过程之前的30gmPEG-PDLLA加入单颈烧瓶中并在80℃下溶解。在磁棒搅拌下,将反应器连接到真空泵,并且在1托或更低的压力下通过升华方法纯化产物24小时和48小时。制备例3:通过升华方法纯化二嵌段共聚物(mPEG-PDLLA)除了纯化温度为100℃以外,通过与制备例2相同的方法进行纯化。制备例4:通过升华方法纯化二嵌段共聚物(mPEG-PDLLA)除了纯化温度为120℃以外,通过与制备例2相同的方法进行纯化。制备例5:使用氧化铝(Al2O3)通过吸附法纯化二嵌段共聚物(mPEG-PDLLA)将在制备例1的聚合反应过程中获得的进行纯化过程之前的30gmPEG-PDLLA加入单颈烧瓶中并加入丙酮(60ml)溶解。向其中加入氧化铝(15g)并完全混合。将单颈烧瓶与旋转蒸发器连接,内容物在50℃、60rpm下混合2小时。然后将溶液在室温下用PTFE滤纸(1μm)过滤以除去氧化铝。使用旋转蒸发器在60℃真空下蒸馏经过滤的丙酮溶液以除去丙酮,由此获得纯化的mPEG-PDLLA。通过用1H-NMR分析获得关于单甲氧基聚乙二醇的末端基团-OCH3的适当峰的相对强度,从而计算分子量(Mn:~3690)。根据上述制备例2-5中的纯化条件的mPEG-PDLLA的分子量变化示于下表1中。表1从表1的结果可以看出,随着纯化温度变高,mPEG-PDLLA的分子量的减少量增加。80-100℃和24-48小时,特别是100℃和24小时的纯化条件可被认为是有效的。比较例1:含有紫杉醇的聚合物胶束组合物的制备称量1g紫杉醇和5g制备例1中得到的mPEG-PDLLA,加入4ml乙醇,在60℃下搅拌直至混合物完全溶解形成澄清溶液。然后使用配备有圆底烧瓶的旋转蒸发器在60℃下减压蒸馏3小时以除去乙醇。然后将温度降低至50℃,加入140ml室温下的蒸馏水,并进行反应直至溶液变成澄清的蓝色以形成聚合物胶束。向其中加入2.5g无水乳糖作为冻干助剂并使其完全溶解,使用孔径为200nm的过滤器过滤,并冷冻干燥,得到含有紫杉醇的粉末状聚合物胶束组合物。实施例1:含有紫杉醇的聚合物胶束组合物的制备除了使用在制备例3中纯化24小时的mPEG-PDLLA之外,通过与比较例1相同的方法制备含有紫杉醇的聚合物胶束组合物。实施例2:含有紫杉醇的聚合物胶束组合物的制备除了使用在制备例5中纯化的mPEG-PDLLA以外,通过与比较例1相同的方法制备含有紫杉醇的聚合物胶束组合物。实验例1:通过液相色谱分离相关化合物向容纳有已经过6个月加速试验(温度:40℃)的100mg含有紫杉醇的聚合物胶束组合物的小瓶中加入16.7ml去离子水(DW),使内容物完全溶解,取总量的液体并转移到20ml容量瓶中,稀释至标线使总体积为20ml(5.0mg/ml)。取2ml该液体并转移到10ml容量瓶中,加入乙腈至标线使总体积为10ml(1mg/ml)。对于上述组合物,使用以下液相色谱法分离相关化合物并分段收集。液相色谱条件1)柱:Poroshell120PFP(4.6×150mm,2.7μm,Agilent)2)流动相:A:DW/B:乙腈时间(min)A%B%0.00653525.00455528.00455530.00653535.0065353)流速:0.6ml/min(分钟)4)注射量:10μl5)检测器:紫外吸收分光光度计(测量波长:227nm)得到的HPLC分析的色谱图显示在图1中。实验例2:使用LC/MS/MS对相关化合物进行定性分析通过液相色谱-质谱仪(LC/MS/MS)的MS扫描和产物离子扫描对实验例1中分离的相关化合物(RRT:0.87±0.02(0.85~0.89))进行定性分析。在下面的测量中,使用液相色谱1200系列和电喷雾电离质谱仪6400系列(Agilent,美国)作为LC/MS/MS。分析条件如下。液相色谱条件1)柱:Poroshell120PFP(4.6×150mm,2.7μm,Agilent)2)流动相:A:DW/B:乙腈时间(min)A%B%0.00653525.00455528.00455530.00653535.0065353)流速:0.6ml/min4)注射量:10μl5)检测器:紫外吸收分光光度计(测量波长:227nm)电喷雾电离质谱仪的条件1)电离:电喷雾电离,正(ESI+)2)MS方法:MS2扫描/产物离子扫描3)离子源:AgilentJetStreamESI4)雾化器气体(压力):氮气(35psi)5)离子喷雾电压:3500V6)干燥气体温度(流速):350℃℃(7L/min)7)鞘气温度(流速):400℃(10L/min)8)碎裂电压:135V9)喷嘴电压:500V10)池加速电压:7V11)EMV:0V12)碰撞能量:22V13)前体离子:m/z894.214)质量扫描范围:m/z100~1500将从检测阶段分离并产生的用于分析的物质设置为在质谱仪中流动,同时定性分析相关化合物的检测离子,并选择质谱的特征离子[M+Na]。实验例3:对酸处理的紫杉醇在RRT0.87下获得的物质进行LC/MS/MS分析作为实验例2中的定性分析的结果,在含有紫杉醇的聚合物胶束组合物的HPLC分析中出现在RRT0.87处的物质被认为是当紫杉醇存在于酸性条件下时产生的物质。为了证实这一点,通过向紫杉醇中加入1NHCl制备化合物,通过使用制备型LC,分离并分段收集了RRT0.87处的物质,并通过LC/MS/MS对其进行分析。LC/MS/MS分析的结果显示在图3中,LC/MS/MS分析中的产物离子扫描结果与经六个月加速试验的聚合物胶束组合物样品的结果一起显示在图4中。根据分析结果,通过用酸处理紫杉醇在RRT0.87处获得的物质显示与相关化合物(RRT:0.87±0.02(0.85~0.89))的HPLC峰在完全相同位置的HPLC峰,所述相关化合物为经过六个月加速试验的本发明的含有紫杉醇的聚合物胶束组合物中的相关化合物。此外,由于这些峰的MS产物离子扫描显示相同的图案,因此可以确认两种物质具有相同的结构。实验例4:酸处理的紫杉醇在RRT0.87处获得的物质的NMR分析将通过向紫杉醇中加入1NHCl制备的化合物分离并在RRT0.87处分段收集,通过NMR分析其结构。在NMR分析中,1HNMR分析的结果显示在图5中,13CNMR分析的结果显示在图6中,COSY(相关光谱)分析的结果显示在图7中,HMBC(异核多键相关光谱)分析示于图8中。根据分析结果,可以确认通过用酸处理紫杉醇在RRT0.87处获得的物质(即经过六个月加速试验的本发明实施方案的含有紫杉醇的聚合物胶束组合物中的相关化合物(RRT:0.87±0.02(0.85~0.89))是紫杉醇和水的以下组合形式的化合物。紫杉醇和一分子水的组合形式:C47H53NO15(871.94g/mol)。核磁共振光谱的条件1.1HNMR1)NMR设备:装备有温度控制器的BruckerDRX-300,AVANCE-4002)样品/溶剂:在外径为5mm的NMR管中的1-10mg样品/0.6mL氯仿-d(在所有NMR实验中,使用相同的样品)3)探头:Brucker5mmQNP或C-H双探头4)质子90°脉冲宽度/激发角/捕获时间:10.0-11.0微秒/30°/~5.0秒5)弛豫延迟/扫描次数:1.0~5.0秒/4~642.13CNMR1)探头:Brucker5mmQNP或C-H双探头2)碳90°脉冲宽度/激发角/捕获时间:10.0微秒/30°/2.0~3.0秒3)弛豫延迟/扫描次数:2.0~5.0秒/大于5,0003.COSY1)NMR设备:BruckerAVANCE-4002)探头:Brucker5mmQNP3)脉冲序列:cosygp脉冲序列4)质子90°脉冲宽度/捕获时间:11.0微秒/0.4~1.6秒5)弛豫延迟/扫描次数/ω1的实验次数:0.5~2.0秒/32~48/400~5004.HMBC1)NMR设备:BruckerAVANCE-4002)探头:Brucker5mm反向探头3)脉冲序列:inv4gptp脉冲序列(对于HMQC)/inv4gplprnd脉冲序列(对于HMBC)4)质子90°脉冲宽度/碳90°脉冲宽度/获取时间:7.5微秒/17~18微秒/0.15~0.2秒5)弛豫延迟/HMQC的扫描次数/HMBC的扫描次数/ω1的实验次数:1.0~2.0秒/32~96/224/128~2566)温度/1/2(JCH):300K/3.5毫秒实验例5:在严格条件下(80℃)含有药物的聚合物胶束的储存稳定性的对比试验将在比较例1和实施例1-2中制备的紫杉醇的聚合物胶束组合物在80℃的烘箱中保持3周,然后用HPLC分析组合物以比较相关化合物的量。通过将胶束组合物溶解在80%乙腈水溶液中并稀释至紫杉醇浓度为600ppm来制备测试溶液。HPLC分析所得的色谱图显示在图9中,并且按照严格试验时间的相关化合物的含量变化(%)示于下表2中。HPLC条件柱:直径2.7μm,Poroshell120PFP(4.6×150mm,2.7μm)(Agilent柱)流动相时间(min)水:乙腈0~2565:35→45:5525~2845:5528~3045:55→65:3530~3565:35检测器:紫外吸收分光光度计(227nm)流速:0.6ml/min各相关化合物的量(%)=100(Ri/Ru)Ri:在测试溶液分析中检测到的每种相关化合物的面积Ru:在测试溶液分析中检测到的所有峰面积的总和[表2]*RRT0.87±0.02:紫杉醇,氧杂环丁烷开环化合物RRT0.96±0.02:紫杉醇,氧杂环丁烷开环化合物RRT1.00:紫杉醇RRT1.10±0.02:紫杉醇,L-丙交酯反应化合物RRT1.12±0.02:紫杉醇,D-丙交酯反应化合物RRT1.44±0.05:紫杉醇,去水化合物从表2和图9可知,与比较例1的组合物相比,实施例1或2的聚合物胶束药物组合物的稳定性得到提高,紫杉醇量的减少相对较小,因此可以更稳定地维持组合物中所含药物的效果。实验例6:在加速条件下(40℃)含有药物的聚合物胶束的储存稳定性的对比试验除了将分别在比较例1和实施例1中制备的紫杉醇的聚合物胶束组合物在稳定性试验仪中于40℃下保持6个月之外,通过与实验例5相同的方法进行实验。将按照加速试验时间的相关化合物的含量变化示于下表3中。[表3]上述试验结果显示了对不同批次的3种或更多种聚合物胶束组合物进行的试验中每种相关化合物和紫杉醇的量的平均值。通过实验例6,已经证明当在加速储存温度下(40℃)储存6个月时,实施例1的聚合物胶束药物组合物具有比比较例1的组合物更低量的相关化合物。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1