用于治疗神经疾病的方法和组合物与流程
本申请要求2016年1月15日提交的美国临时申请号62/279,394;2016年2月26日提交的美国临时号62/300,271;和2016年4月28日提交的美国临时号62/328,925的权益,其公开内容据此全文以引用方式并入。
本公开涉及神经疾病,并且尤其是具有中枢神经系统障碍的溶酶体疾病,并且包括i型粘多糖贮积病(mpsi),和“减毒形式”诸如scheie综合征(mpsis)和hurler-scheie综合征(mpsh-s),以及更快速进展形式hurler综合征(mpsih)和ii型粘多糖贮积病(mpsii)两者,也称为hunter综合征的治疗以及基因疗法的领域。
背景技术:
基因疗法为人类治疗的新时代提供了巨大的潜力。这些方法将允许治疗迄今为止尚未通过标准医学实践解决的病状。特别有希望的一个领域是能够将转基因添加到细胞中以使所述细胞表达先前未在所述细胞中产生的产物。此技术的用途的实例包括插入编码治疗性蛋白的基因、插入编码细胞或个体中某种程度上缺少的蛋白质的编码序列以及插入编码结构核酸诸如微rna中的序列。
转基因可通过多种方式递送至细胞,使得转基因整合到细胞自身的基因组中并维持在那里。近年来,已开发了转基因整合策略,其使用用位点特异性核酸酶进行的裂解以便靶向插入选择的基因组基因座中(参见例如,共同拥有的美国专利7,888,121)。核酸酶,诸如锌指核酸酶(zfn)、转录激活因子样效应子核酸酶(talen)或核酸酶系统诸如crispr/cas系统(利用工程化指导rna)对靶基因具有特异性,并且可被利用使得通过同源定向修复(hdr)或通过非同源末端连接(nhej)驱动过程中的末端捕获插入转基因构建体。参见例如,美国专利号9,394,531;9,255,250;9,200,266;9,045,763;9,005,973;9,150,847;8,956,828;8,945,868;8,703,489;8,586,526;6,534,261;6,599,692;6,503,717;6,689,558;7,067,317;7,262,054;7,888,121;7,972,854;7,914,796;7,951,925;8,110,379;8,409,861;美国专利公布20030232410;20050208489;20050026157;20050064474;20060063231;20080159996;201000218264;20120017290;20110265198;20130137104;20130122591;20130177983;20130196373;20150056705和20150335708,所述专利的公开内容以引用方式全文并入。
靶基因座包括“安全港”基因座,诸如人细胞中的aavs1、hprt、白蛋白和ccr5基因,以及鼠细胞中的rosa26。参见例如,美国专利号9,394,545;9,222,105;9,150,847;8,895,264;8,771,985;8,110,379;7,951,925;美国公布号20100218264;20110265198;20150056705和20150159172。与依赖于转基因随机整合的经典整合方法相比,核酸酶介导的整合提供了改善转基因表达、增加安全性和表达耐久性的前景,因为它允许精确的转基因定位以使基因沉默或激活附近癌基因的风险最小化,这进而允许提供用于治疗和/或预防疾病的蛋白质。
虽然将转基因递送至靶细胞是完全实施此技术必须克服的一个障碍,但必须攻克的另一个问题是确保在将转基因插入细胞并表达后,如此编码的基因产物必须达到有机体的必要位置,并在足够的局部浓度下使其有效。对于特征在于缺少蛋白质或存在异常非功能性蛋白质的疾病,转基因编码的野生型蛋白质的递送可能是极其有帮助的。
溶酶体贮积病(lsd)是一组罕见的代谢单基因疾病,其特征在于缺少通常参与废脂质,粘多糖(即糖胺聚糖(gag))分解的功能性单个溶酶体蛋白。这些疾病的特征在于这些化合物在细胞中的积聚,因为由于特定酶的错误功能而不能将它们加工用于再循环。最初认为lsd的病理生理学与gag的简单沉积有关,但目前的研究已得到对这些疾病的复杂性的认识。gag贮积似乎导致细胞、组织和器官稳态的扰动,并且还与导致炎症反应激活的细胞因子和炎症调节剂的分泌增加有关(muenzer(2014)molgenmetabol111:63-72)。
最常见的实例是gaucher病(葡糖脑苷脂酶缺乏症-基因名称:gba)、fabry病(α半乳糖苷酶a缺乏症:gla)、hunter综合征(也称为mpsii,艾杜糖醛酸2-硫酸酯酶缺乏症:ids)、hurler综合征(也称为mpsih,α-l艾杜糖醛酸酶缺乏症:idua)、pompe病(α-葡萄糖苷酶:gaa)以及a型和b型niemann-pick病(鞘磷脂磷酸二酯酶1缺乏症-smpd1)疾病。当将所有分组集中在一起时,lsd在7000名新生儿中的人口中发病率为约1。治疗选择包括酶替代疗法(ert),其中通常通过大剂量静脉内注射向患者提供缺失的酶。这种治疗仅用于治疗症状并且不是治愈性的,因此患者必须在其余生中重复给药这些蛋白质,并且可能产生针对注射蛋白质的中和抗体。
mpsi与idua(α-l艾杜糖醛酸酶)缺乏相关,并且每100,000名新生儿中大约发生一次。idua酶缺乏导致gag在几个器官包括脑的溶酶体中积聚,这导致所述疾病的临床表现。gag可在尿液、血浆和组织中检测到,并且可用作疾病进展的生物标志物。它是一种常染色体隐性遗传病,并且具有一个正常idua基因和一个突变拷贝的杂合子受试者产生减少量的酶,但可能是无症状的,因为由野生型基因产生了足够的酶。对于患者,hurler综合征的症状最经常出现在3至8岁之间,并且可能包括脊柱中的异常骨骼、爪形手、浑浊角膜、耳聋、停止生长、心脏瓣膜问题、关节疾病,包括僵硬、随时间推移变得更糟的智力残疾,以及面部特征厚、粗,具有低鼻梁。甚至患有严重hurler综合征的婴儿在出生时看起来正常,并且在出生后的头两年内面部症状可能变得更加明显。随着孩子年龄的增长,他们的身材矮小(最大为大约4英尺),并且经常在10岁之前死于阻塞性气道疾病、呼吸道感染或心脏并发症。
临床上,mpsi分为三类:hurler综合征、hurler-scheie综合征和scheie综合征,以最初描述病状的医生命名。hurler综合征通常被认为是最严重的并且脑受到影响;scheie综合征是最‘温和’[或减毒]。许多人介于两者之间,并且被称为hurler-scheie。
hunter综合征(ii型粘多糖贮积病,mpsii)是一种罕见的x-连锁溶酶体病症,其由缺少功能性艾杜糖醛酸2-硫酸酯酶(ids)和随后在受影响个体中的糖胺聚糖(gag)积聚引起。hunter综合症每出生100,000至150,000名男性中大约有1名,并且在女性中很少发生。mpsii是一种可变的、进行性的多系统病症,其中在大多数患者中,症状严重并且在早龄期(10-20岁之间)发生死亡,尽管在患有所述病症的更减毒形式的患者中,已报道了存活到50和60岁(wraith等(2008)eurjpediatr167:267-277)。
临床上,所述系列最末端的患者在出生时看起来正常,但经常基于如复发性呼吸道感染、发育迟缓、关节僵硬和髋关节发育不良、复发性脐带和腹股沟疝的症状,以及与mpsii相关的典型面部特征的发展(突出的前额、具有扁平鼻梁的鼻子、扩大的舌头和扩大的头部)在18至36个月之间诊断出来。随着疾病的进展,临床表现变得多系统,从而影响肺和心脏系统以及肌肉骨骼系统和cns。严重与减毒形式之间的差异主要是由于神经变性和精神损伤的存在。
mpsi和mpsii的标准治疗是酶替代疗法(ert),从100,000美元至500,000美元不等,或造血干细胞移植(hsct),单轮花费大约200,000美元,或两者的组合。然而,这些治疗选择具有实质性缺点。对于ert,一些长期治疗研究显示,ert使用持续时间与生活质量恶化之间存在统计学显著相关性(aronovich和hackett(2015)molgenmetabol114:83-93)。另外,ert用于改善躯体疾病(不涉及cns),并且对于治疗神经疾病是低效的,因为外源性给予的酶不穿过血脑屏障(muenzer同上)。对于hsct,存在由hsct过程导致的显著死亡率(至少10%)(aronovich和hackett,同上)。因此,目前可用的治疗使得具有cns损伤的最严重的mpsi和mpsii患者得不到充分服务。
因此,仍需要可用于治疗mpsi和mpsii疾病的方法和组合物,包括通过基因组编辑进行治疗,例如,递送编码基因产物的转基因,使得转基因的表达将处于治疗相关水平。
技术实现要素:
本文公开了用于治疗和/或预防hurler(或mpsi)疾病以及用于治疗和/或预防hunter(mpsii)综合征的方法和组合物。本发明描述了将转基因序列插入合适的靶细胞中的方法,其中所述转基因编码治疗疾病的蛋白质(例如,用于mpsi的全长或截短的idua蛋白;用于mpsii的全长或截短的ids蛋白),例如在mpsi或mpsii中缺少或缺乏的蛋白质,其在由idua或ids转基因提供时分别治疗和/或预防mpsi或mpsii。本发明还描述了用表达系统转染和/或转导合适的靶细胞的方法,使得编码转基因的idua或ids表达治疗疾病(例如,减轻与疾病相关的一种或多种症状)的蛋白质。idua或ids蛋白可从靶细胞中排出,使得它能够影响不携带转基因的其它细胞或被其摄取(旁观者效应或交叉校正)。本发明还提供了用于生产产生高水平idua或ids的细胞(例如,成熟或未分化细胞)的方法,其中将这些改变的细胞群引入患者体内将提供所需的蛋白质以治疗和/或预防mpsi或mpsii。另外,本发明提供了用于生产产生idua或ids的高活性形式(治疗性)的细胞(例如成熟或未分化细胞)的方法,其中将这些改变的细胞群引入患者体内或在患者体内形成这些改变的细胞群将提供所需的蛋白质活性以治疗mpsi或mpsii疾病(例如,减轻或消除一种或多种症状)。
在一个方面,本文提供了一种减少或预防患有i型粘多糖贮积病(mpsi)和/或i型粘多糖贮积病(mpsii)的受试者的中枢神经系统(cns)损伤的方法,所述方法包括向受试者施用无启动子供体载体,其包含编码人α-l-艾杜糖醛酸酶(hidua)和/或人艾杜糖醛酸-2-硫酸酯酶(hids)的转基因;以及施用编码靶向内源白蛋白基因的一对锌指核酸酶(zfn)的表达载体,其中转基因在裂解肝细胞中的白蛋白基因后整合到白蛋白基因中,使得编码的hidua和/或hids由肝脏表达和分泌,并且进一步地,其中由肝脏分泌的hidua和/或hids在受试者的中枢神经系统(cns)中发现(例如,穿过血脑屏障),使得cns损伤可被减少或预防。在本文所述的任何方法中,cns损伤包括认知缺陷(例如,与未治疗的个体相比,学习能力丧失的降低);转基因调节受试者脑中糖胺聚糖的水平;和/或在cns(脑和/或脊髓)中减少或消除受试者中的神经元或神经胶质细胞空泡化。所述方法还可包括在施用供体载体和表达载体之前和之后向受试者施用免疫抑制剂。在本文所述的任何方法中,所述zfn对包含如表1所示的锌指蛋白。zfn表达载体和/或供体载体可以是腺相关载体(aav),其可通过任何方法施用,包括通过静脉内注射。在某些实施方案中,向受试者施用的zfn:zfn:供体比率是1:1:8。
在另一方面,本发明描述了一种在受试者的细胞中表达编码一种或多种校正性idua或ids转基因的转基因的方法。转基因可插入合适的靶细胞(例如,血细胞、肝细胞、脑细胞、干细胞、前体细胞等)的基因组中,使得由所述校正性转基因编码的idua或ids产物稳定地整合到细胞的基因组中或者可替代地,转基因可在染色体外维持在细胞中。在一个实施方案中,将校正性idua或ids转基因(稳定地或染色体外)插入细胞系中用于体外产生idua或ids,然后可向受试者施用所述(任选地纯化和/或分离的)蛋白质用于(例如,通过减少和/或消除与mpsi或mpsii疾病相关的一种或多种症状)治疗患有mpsi或mpsii疾病的受试者。
因此,在另一方面,本文描述了(例如,通过减少和/或消除与mpsi或mpsii疾病相关联的一种或多种症状)治疗患有mpsi或mpsii疾病的受试者的离体或体内方法,所述方法包括向有需要的受试者提供idua或ids蛋白。在某些实施方案中,所述方法包括体内方法,例如将idua或ids转基因插入如本文所述的细胞中,使得蛋白质在患有mpsi或mpsii疾病的受试者中产生。在一些实施方案中,表达的活性表达的idua和/或ids达到患有mpsi或mpsii疾病的受试者中的内源水平的1、2、3、4、5、10、15、20、25、30、40、50、60、70、80、90、100、200、300或更大百分比。在一些实施方案中,表达的活性idua或ids酶达到在野生型个体中发现的酶水平的1、2、3、4、5、10、15、20、25、30、40、50、60、70、80、90、100、200、300或更大百分比。在其它实施方案中,包含idua或ids转基因的分离的细胞可用于例如,通过向患有mpsi或mpsii疾病的受试者施用所述细胞来治疗有需要的患者。在其它实施方案中,将校正性idua或ids转基因和/或表达idua或ids的细胞插入有需要的受试者中,使得替代蛋白质在体内产生。在一些优选的实施方案中,将校正性转基因插入受试者细胞的基因组中,而在其它优选的实施方案中,将校正性转基因插入受试者的细胞中并在染色体外维持在细胞中。转基因可整合到多种靶组织中,包括肝脏、脑、肌肉、心脏、肺等。在本文所述的任何方法中,表达的idua或ids蛋白可从细胞中排出(例如通过输出到血液中),以作用于缺少idua或ids转基因的其它细胞或被其摄取(例如通过受体介导的内吞作用通过甘露糖受体或甘露糖-6-磷酸受体)。因此,在某些实施方案中,表达idua或ids转基因的细胞(例如,肝脏、hspc或源自编辑的hspc的细胞)分泌idua或ids蛋白,其然后作用于一种或多种次级组织(例如,脑、肌肉、骨骼、心脏、肺、脾脏等)。在一些情况下,靶组织(其表达idua或ids转基因)是肝脏。在其它情况下,靶组织是脑。在其它情况下,靶标是血液(例如,脉管系统)。在其它情况下,靶标是骨骼肌。还在其它情况下,靶组织是显示疾病病理生理学的另外方面的那些(例如心脏瓣膜、骨、关节组织、气道组织等)。
在本文所述的任何方法中,校正性idua或ids基因包含功能性idua或ids基因的野生型序列(包括野生型的功能片段),而在其它实施方案中,以某种方式改变校正性idua或ids转基因的序列,例如以给出增强的生物活性(例如,优化的密码子以增加生物活性和/或截短idua或ids编码序列)。在优选的实施方案中,本发明的方法和组合物在有需要的mpsi或mpsii受试者中产生活性idua或ids酶。表达的idua和/或ids酶能够降解糖胺聚糖(gag)。在一些实施方案中,gag在身体的特定组织中减少,包括肝脏、脾脏、肾脏、肺、心脏、肌肉和脑。在优选的实施方案中,与未治疗的mpsi或mpsii受试者相比,任何组织中的gag水平降低1%、2%、3%、4%、5%、10%、15%、20%、25%、30%、40%、50%、60%、70%、80%、90或100%或其间的任何值。
在本文所述的任何方法中,可使用核酸酶将idua或ids转基因插入靶细胞的基因组中。合适的核酸酶的非限制性实例包括锌指核酸酶(zfn)、talen(转录激活因子如蛋白质核酸酶)和/或crispr/cas核酸酶系统,其包括与细胞基因组中的感兴趣区域中的靶位点(例如,疾病相关基因、高度表达的基因、白蛋白基因或其它或安全港基因)结合的dna结合分子,以及一个或多个核酸酶结构域(例如,裂解结构域和/或裂解半结构域)。裂解结构域和裂解半结构域可例如从各种限制性内切核酸酶、cas蛋白(1类或2类)和/或归巢内切核酸酶获得。在某些实施方案中,锌指结构域识别白蛋白基因中的靶位点或红细胞(rbc)中的珠蛋白基因。参见例如,美国公布号2014001721,其以引用方式全文并入本文。在其它实施方案中,zfn、talen和/或crispr/cas系统结合和/或裂解安全港基因,例如ccr5基因、ppp1r12c(也称为aavs1)基因、白蛋白、hprt或rosa基因。参见例如,美国专利号7,888,121;7,972,854;7,914,796;7,951,925;8,110,379;8,409,861;8,586,526;美国专利公布20030232410;20050208489;20050026157;20060063231;20080159996;201000218264;20120017290;20110265198;20130137104;20130122591;20130177983;20130177960和20140017212。核酸酶(或其组分)可作为编码本文所述的一种或多种zfn、talen和/或crispr/cas系统的多核苷酸提供。多核苷酸可以是例如mrna。在一些方面,mrna可以是化学修饰的(参见例如kormann等,(2011)naturebiotechnology29(2):154-157)。在其它方面,mrna可包含arca帽(参见美国专利7,074,596和8,153,773)。在进一步的实施方案中,mrna可包含未修饰和修饰的核苷酸的混合物(参见美国专利公布20120195936)。在仍进一步的实施方案中,mrna可包含wpre元件(参见美国专利申请62/158,277)。
在另一方面,本发明提供了一种工程化核酸酶蛋白,其能够裂解(编辑)干细胞或前体细胞(例如,血细胞前体、肝干细胞等)的基因组以便引入希望的idua或ids转基因。在一些方面,然后扩增编辑的干细胞或前体细胞,并且可诱导其离体分化成成熟的编辑细胞,且然后将所述细胞给予患者。在其它方面,编辑的前体(例如,cd34+干细胞)在骨髓移植中给予,其在成功植入后增殖产生编辑的细胞,然后在体内分化和成熟并含有由idua或ids转基因表达的生物制剂。在其它方面,编辑的干细胞是肌肉干细胞,然后将其引入肌肉组织中。在一些方面,工程化核酸酶是锌指核酸酶(zfn)并且在其它方面,核酸酶是tale核酸酶(talen),并且在其它方面,使用crispr/cas系统。可将核酸酶工程化以对安全港基因座、与疾病相关的基因或在细胞中高度表达的基因具有特异性。仅作为非限制性实例,安全港基因座可以是aavs1位点、ccr5基因、白蛋白或hprt基因,而疾病相关基因可以是idua或ids基因。
在本文所述的任何方法中,核酸酶(例如,zfn、talen和/或crispr/cas系统)可以多核苷酸形式提供,例如包含编码一种或多种核酸酶(或其组分)的多核苷酸的表达载体,其任选地可操作地连接至启动子。在一个实施方案中,表达载体是病毒载体。在另一方面,本文所述的是idua或ids表达载体,其包含任选地可操作地连接至启动子的多核苷酸,所述多核苷酸编码如本文所述的idua或ids。在一个实施方案中,表达是病毒载体。
在另一方面,本文所述的是一种宿主细胞,其包含一种或多种如本文所述的zfn、talen和/或crispr/cas系统表达载体和/或idua或ids表达载体。宿主细胞可与一种或多种zfp、talen和/或crispr/cas系统表达载体稳定转化或瞬间转染或其组合。在一些实施方案中,宿主细胞是肝细胞。因此,本文提供了一种hep2g细胞,其包含编码整合到白蛋白基因的内含子1中的hiuda和/或hids的无启动子供体载体,其中hiuda和/或hids由hep2g细胞表达和分泌。还提供了一种包含如权利要求11所述的hep2g细胞的细胞培养物(其中所述培养基包含hiuda或hids),如本文所述其本身是一种包含由细胞分泌和/或从细胞的培养基分离的hiuda和/或hids的药物组合物。
在其它实施方案中,提供了用如本文所述的治疗性idua或ids转基因替代任何靶基因中的基因组序列的方法,例如使用如本文所述的一种或多种核酸酶(例如,zfn、talen和/或crispr/cas系统(或编码所述zfn、talen和/或crispr/cas系统的载体)和在用zfn、talen和/或crispr/cas系统靶向裂解后插入基因中的“供体”序列或idua或ids转基因。供体idua或ids序列可存在于携带核酸酶(或其组分)的载体中、存在于单独的载体(例如,ad、aav或lv载体或mrna)中或者可替代地,可使用不同的核酸递送机制引入细胞中。将供体核苷酸序列插入靶基因座(例如,高度表达的基因、疾病相关基因、其它安全港基因等)中导致在靶基因座(例如,白蛋白、球蛋白等)的内源遗传控制元件控制下表达idua或ids转基因。在一些方面,将idua或ids转基因插入例如靶基因(例如,白蛋白)中导致完整idua或ids蛋白序列的表达,并且缺少由靶标编码的任何氨基酸(例如,白蛋白)。在其它方面,表达的外源idua或ids蛋白是融合蛋白,并且包含由idua或ids转基因和由idua或ids转基因插入其中的内源基因座编码的氨基酸(例如,来自内源靶基因座或者可替代地,来自转基因上编码靶基因座序列的序列)。靶标可以是任何基因,例如安全港基因,诸如白蛋白基因、aavs1基因、hprt基因;ccr5基因;或高度表达的基因,诸如rbc中的珠蛋白基因(例如,β珠蛋白或γ珠蛋白)。在一些情况下,内源序列将存在于外源idua蛋白的氨基(n)-末端部分上,而在其它情况下,内源序列将存在于外源idua或ids蛋白的羧基(c)-末端部分上。在其它情况下,内源序列将存在于idua外源蛋白的n-和c-末端部分两者上。内源序列可包括全长野生型或突变体内源序列或者可替代地,可包括部分内源氨基酸序列。在一些实施方案中,内源基因-转基因融合体位于细胞内的内源基因座处,而在其它实施方案中,内源序列-转基因编码序列插入基因组内的另一个基因座中(例如,idua或ids-转基因序列插入白蛋白、hprt或ccr5基因座中)。在一些实施方案中,表达idua或ids转基因使得治疗性idua或ids蛋白产物保留在细胞(例如,前体或成熟细胞)内。在其它实施方案中,idua或ids转基因与膜蛋白的细胞外结构域融合,使得在表达后,转基因idua或ids融合将导致治疗性蛋白的表面定位。参见例如,美国专利公布号20140017212。在一些方面,编辑的细胞还包含跨膜蛋白以将细胞运输至特定组织类型。在一个方面,跨膜蛋白是抗体,而在其它方面,跨膜蛋白是受体。在某些实施方案中,细胞是前体(例如,cd34+或造血干细胞)或成熟rbc。在一些方面,在转基因上编码的治疗性idua或ids蛋白产物被输出到细胞外以影响缺少转基因的细胞或被其摄取。在某些实施方案中,细胞是肝细胞,其将治疗性idua或ids蛋白释放到血流中以作用于远端组织(例如,脑、心脏、肌肉等)。
本发明还提供了用于产生携带idua或ids治疗性蛋白的细胞(例如,rbc)的方法和组合物以便治疗mpsi疾病,其可作为同种异体产品普遍用于所有患者。这些rbc载体可包含跨膜蛋白以帮助细胞的运输。在一个方面,跨膜蛋白是抗体,而在其它方面,跨膜蛋白是受体。
在一个实施方案中,idua或ids转基因在插入白蛋白基因座中后由白蛋白启动子表达。如果转基因在体内插入肝细胞中,则由idua或ids转基因编码的生物制剂然后可释放到血流中。在一些方面,idua或ids转基因通过静脉内或其它注射在体内在病毒载体中递送至肝脏。
在一些实施方案中,将idua或ids转基因供体转染或转导到细胞中用于转基因的游离型或染色体外维持。在一些方面,将idua或ids转基因供体维持在包含调节结构域的载体上以调节转基因供体的表达。在其它方面,将包含转基因供体的载体在体内递送至合适的靶细胞,使得如果转基因供体载体递送至肝细胞,则由转基因供体编码的idua或ids治疗性蛋白释放到血流中。
在另一个实施方案中,本发明描述了已插入idua或ids转基因的前体细胞(造血干细胞、肌肉干细胞或cd34+造血干细胞(hsc)细胞),使得源自这些前体的成熟细胞含有高水平的由转基因编码的idua或ids产物。在一些实施方案中,这些前体是诱导多能干细胞(ipsc)。
在一些实施方案中,本发明的方法可在转基因动物系统中体内使用。在一些方面,转基因动物可用于模型开发,其中转基因编码人idua或ids蛋白。在一些情况下,转基因动物可在相应的内源基因座处敲除,从而允许开发体内系统,其中可分离研究人蛋白质。此类转基因模型可用于筛选目的以鉴定小分子、或大生物分子或可与感兴趣人蛋白质相互作用或修饰其的其它实体。在一些方面,将idua或ids转基因整合到选定的基因座(例如,高度表达或安全港)中,整合到通过本文所述的任何方法获得的干细胞(例如,胚胎干细胞、诱导多能干细胞、肝干细胞、神经干细胞等)或非人动物胚胎中,并且然后将胚胎植入使得活的动物出生。然后将动物饲养至性成熟并使其产生后代,其中至少一些后代包含整合的idua或ids转基因。
在又一个方面,本文提供了一种用于将核酸序列位点特异性整合到染色体(例如整合到非人类胚胎的染色体中)的内源基因座(例如,疾病相关的、高度表达的,诸如肝细胞中的白蛋白基因座或rbc中的珠蛋白基因座)中的方法。在某些实施方案中,所述方法包括:(a)给非人胚胎注入(i)至少一种dna载体,其中所述dna载体包含位于待整合的idua或ids编码核酸序列侧面的上游序列和下游序列,和(ii)至少一种编码识别靶基因座中的整合位点的锌指、tale核酸酶或crispr/cas系统的rna分子,以及(b)培养胚胎以使zfn、talen和/或crispr/cas系统表达,其中通过使用dna载体进行的同源重组来修复通过zfn、talen和/或crispr/cas系统引入整合位点中的双链断裂,以便将核酸序列整合到染色体中。
在任何先前的实施方案中,本发明的方法和化合物可与其它治疗剂组合用于治疗患有mpsi或mpsii疾病的受试者。在一些方面,所述方法和组合物与允许穿过血脑屏障的方法和组合物组合使用。在其它方面,所述方法和组合物与已知抑制受试者免疫应答的化合物组合使用。
还提供了一种包含如本文所述的核酸酶系统和/或idua或ids供体的试剂盒。试剂盒可包含编码zfn、talen和/或crispr/cas系统的核酸(例如rna分子或包含在合适表达载体中的zfn、talen和/或crispr/cas系统编码基因)、供体分子、编码单指导rna合适宿主细胞系的表达载体、用于进行本发明方法的说明书等。
鉴于作为整体的公开内容,这些和其它方面对于技术人员将是显而易见的。
附图说明
图1a和1b描绘了用小鼠白蛋白特异性zfn和idua供体治疗指定小鼠组后的结果。在图1a中,图指示在给药后1个月(左图)和4个月(右图)用raav2/8小鼠替代zfn和hidua供体治疗的小鼠肝脏中zfn活性水平(插入缺失%)。从个体小鼠的肝脏中分离基因组dna,并通过在zfn靶位点处对小鼠白蛋白基因座进行深度测序来测量zfn活性。列出研究组编号,并且各个体符号表示在所述时间点(尸检时)来自个体小鼠的数据。水平线表示各组的平均值±标准偏差。在图1b中,通过蛋白质印迹分析测量idua表达。将替代小鼠zfn和hidua供体包装为raav2/8并iv注射到小鼠中。在给药后1个月(图i)或4个月(图ii)对小鼠实施安乐死,并从肝脏中提取蛋白质。使用人特异性idua抗体测定hidua在小鼠肝脏中的表达。gapdh示出为上样对照。
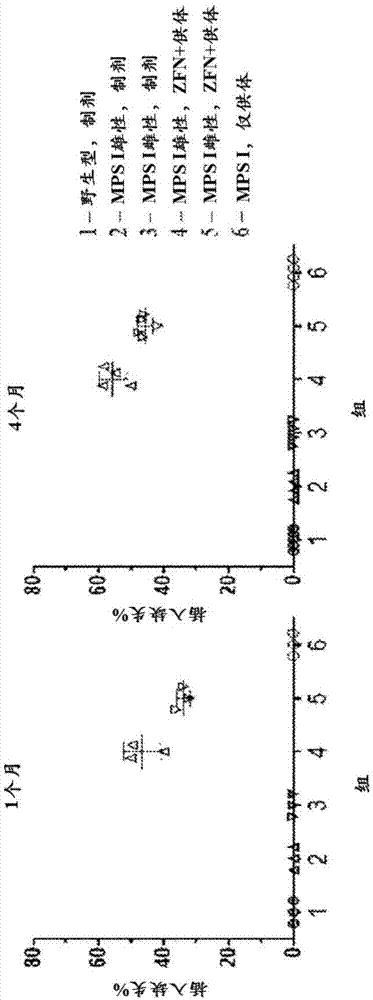
图2a至2c描绘了可检测的idua酶活性。图2a是描绘血浆中酶活性的图。将替代小鼠zfn和hidua供体包装为raav2/8并iv注射到小鼠中。在指定的天数收获血浆,并通过荧光测定法测定idua酶活性。数据示出4-8只动物/组的平均值±sd,取决于时间点。双向重复测量anova随后进行dunnett多重比较测试显示出,mpsizfn+供体组对于雄性动物从第14-120天与野生型组显著不同,并且对于雌性动物从第28-120天显著不同。图2b示出了在肝脏(左图)、血浆(中图)、脾脏(右图)、肾脏(右图)和肺(右图)中直至第60天测量的idua活性。在描绘肝脏、脾脏、肾脏和肺数据的图中,最左边的条(1)表示mpsi杂合子小鼠,右边的下一个条(2)表示未治疗的mpsi纯合子小鼠,右边的下一个条(3)表示仅用idua供体aav治疗的mpsi小鼠,并且右边最远的条(4)表示来自用供体aav和包含白蛋白特异性zfn的aav治疗的mpsi小鼠的数据。图2c具有两个直方图,其描绘了在1个月(图(i))或4个月(图(ii))处死的小鼠的各种组织中idua活性的量。示出的组是:(1-最左边的条)野生型小鼠,给予制剂缓冲液;(2-左边第二个条)mpsi雄性,制剂缓冲液;(3-左边第三个条)mpsi雌性,制剂缓冲液;(4-右边第三个条)mpsi雄性,zfn+供体;(5-右边第二个条)mpsi雌性,zfn+供体;(6-最右边的条)mpsi,仅供体。
图3a至3c是描绘治疗后治疗小鼠中gag水平显著降低的图。将替代小鼠zfn和hidua供体包装为raav2/8并iv注射到小鼠中。在指定的天数收集尿液,并通过blyscangag测定法测定gag水平。图3a示出了治疗后尿液中的gag结果。各组最左边的条示出未治疗的mpsi雄性;左边第二个条示出未治疗的mpsi雌性;右边第二个条示出治疗的mpsi雄性,并且最右边的条示出治疗的mpsi雌性。数据示出1-8只动物/组的平均值±sd,取决于时间点。图3b示出在治疗后1个月(图3b(i))或治疗后4个月(图3b(ii))在各种器官中检测到的gag的量。示出的组与如上图2b和2c中的相同。图3c是示出在治疗后120天内各组尿液中检测到的gag量的图。治疗组与图3b中的相同。
图4a至4f示出了一系列图,其描绘了在来自治疗小鼠的不同组织中检测到的idua酶活性的量。将替代小鼠zfn和hidua供体包装为raav2/8并iv注射到小鼠中。在给药后4个月对小鼠实施安乐死,并从肝脏和其它组织中提取蛋白质。通过荧光测定法测定idua酶活性。数据示出8只动物/组的平均值±sd。*p<0.05相对于各自对照组(制剂治疗的mpsi小鼠)(mann-whitney检验)。图4a示出了所示组中肝脏中的idua活性。图4b示出了所示组中脾脏中的idua活性。图4c示出了所示组中肺中的idua活性。图4d示出了肌肉中的idua活性。图4e示出了所示组中脑中的idua活性,并且图4f示出了所示组中心脏中的idua活性。
图5a至5f是描绘治疗后治疗小鼠中gag水平显著降低的图。将替代小鼠zfn和hidua供体包装为raav2/8并iv注射到小鼠中。在给药后1和4个月对小鼠实施安乐死,并从肝脏和其它组织中提取蛋白质。通过荧光测定法测定idua酶活性。数据示出8只动物/组的平均值±sd。*p<0.05相对于各自对照组(制剂治疗的mpsi小鼠)(mann-whitney检验)。图5a示出了所示组中肝脏中的gag水平。图5b示出了所示组中脾脏中的gag水平。图5c示出了所示组中肺中的gag水平。图5d示出了肌肉中的gag水平。图5e示出了所示组中脑中的gag水平,并且图5f示出了所示组中心脏中的gag水平。
图6a至6d是一系列照片的复印件,其示出了测量给药后4个月的认知表现的巴恩斯迷宫(barnesmaze)。巴恩斯迷宫包括一个带40个孔的平台(图6a)。将小鼠置于平台上并从上方用明亮的光照进行拍摄(图6b)。所有孔都被阻塞(图6c),除了一个是小鼠试图避开光的逃生孔(图6d)。
图7a和7b是描绘在给药后约4个月使用巴恩斯迷宫测试的认知表现结果的图。数据表示在6天的测试(每天平均4次试验)中动物发现目标逃生孔所花费的时间的平均值±sem。图7a示出了mpsi雄性(未治疗)的结果,*p<0.05,**p<0.01,***p<0.001相对于野生型;#p<0.05,###p<0.001相对于mpsi雄性(zfn+供体);并且图7b示出了mpsi雌性(zfn+供体)的结果,*p<0.05相对于野生型;#p<0.05相对于mpsi(仅供体)(双向重复测量anova,随后进行tukey多重比较测试)。
图8a至8c描绘了mpsii小鼠模型中ids的表达。图8a是蛋白质印迹,其示出了在处理后1个月(上图)或4个月(下图)的小鼠肝脏中人ids的表达。使用上样对照检测小鼠gapdh(来自genscript的a00191),针对ids的抗体针对人蛋白质(来自r&dsystems的af2449)而产生。图8b是示出在治疗后直至120天检测到的血浆中ids酶活性的图,并且图8c是示出给药后4个月各种组织中ids酶活性的直方图。图8c是示出各组织的治疗组的分组的图,如下:(1-最左边的条)野生型,未治疗的小鼠;(2-左边第二个条)mpsii小鼠,未治疗;(3-左边第三个条)mpsii小鼠,zfn+供体治疗,低剂量;(4-右边第3个条)mpsii小鼠,zfn+供体治疗,中剂量;(5-右边第二个条)mpsii小鼠,zfn+供体治疗,高剂量;(6-最右边的条)mpsii小鼠,仅供体治疗。组中样品的顺序是从左到右,1-6。指示了数据显著性。
图9a和9b是示出治疗后4个月mpsii小鼠的指定器官中gag水平的直方图。图9a描绘了肝脏、脾脏、肾脏和肺中的水平,而图9b描绘了心脏、肌肉和脑中的水平。星号指示与mpsii未治疗小鼠相比的数据显著性。测试的组和示出的顺序与图8c相同。
图10是示出在治疗后4个月使用巴恩斯迷宫测试的认知表现结果的图。数据表示在6天的测试(每天平均4次试验)中动物发现目标逃生孔所花费的时间的平均值±sem。星号指示在mpsii未治疗小鼠与zfn+供体小鼠之间相比较的数据显著性,并且标签指示在mpsii未治疗小鼠与野生型未治疗小鼠之间相比较的显著性。
图11a至11d是描绘用白蛋白特异性zfn和ids或idua供体转导的hepg2细胞中的ids或idua活性的图。在72小时上清液积聚后,通过hidselisa(图11a中所示)和ids酶活性(图11b中所示)测定分析衍生自用sbs#47171、sbs#47931和sb-ids供体(ids)或单独sbs#47171和sbs#47931(仅zfn)转导的hepg2细胞的四个亚克隆。在72小时上清液积聚后,通过hiduaelisa(图11c中所示)和idua酶活性(图11d中所示)测定分析衍生自用sbs#47171、sbs#47931和sb-idua供体(idua)或单独sbs#47171和sbs#47931(仅zfn)转导的hepg2细胞的三个亚克隆。数据表示三种独立上清液的平均值±sd。
图12a至图12i指示可能在白蛋白基因座处发生的ids供体(“sb-ids”)或idua(“sb-idua”)的整合类型以及在四个ids和三个idua亚克隆中观察到的结果。通过nhej(图12a)或hdr(图12b)在人白蛋白基因座处的sb-ids整合事件用各自的特异性试剂(引物和探针)和结合位点示出。图12c是所有亚克隆的
图13是示出在转导的肝脏中产生ids然后摄取并随后加工成最终成熟蛋白质的过程的示意图。
图14a和14b示出了原代肝细胞中hepg2亚克隆对分泌的ids的摄取。图14a示出了ids蛋白成熟后的不同蛋白质形式,并示出了将hepg2上清液加入到原代肝细胞培养物中的过程。图14b示出了蛋白质印迹,其描绘了在hepg2克隆(“mix”和55)上清液和沉淀中检测到的ids形式,以及在原代肝细胞沉淀中检测到的形式,说明了在产生成熟形式(45kda)的过程中全长多肽(90kda)的加工。还示出了持家蛋白(α-gapdh)的上样对照以及从上清液更新ids后原代肝细胞沉淀中的相对ids活性。
图15a至15d说明了对hepg2亚克隆中产生的ids或idua的糖基化模式的研究。图15a示出了去糖基化中使用的糖苷酶的不同种类的糖基化模式和底物特异性(修改自lee等(2009)natprotoc.4(4):592-604)。图15b和15c是使用抗ids抗体的蛋白质印迹,其研究在hepg2克隆中产生的ids蛋白,其中图15b示出了在细胞上清液中发现的蛋白质,而图15c示出了用糖苷酶png酶f和endoh处理后的细胞沉淀中的蛋白质。用png酶f处理去除了所有寡糖链,这导致如所示的“非糖基化hids”。对于hepg2沉淀蛋白质印迹,hsp90示出为上样对照‘mix’=由2个不同ids亚克隆组成的混合hepg2ids群。‘克隆55’=hepg2ids亚克隆#55。‘对照’=具有idua而不是ids整合在白蛋白基因座处作为阴性对照的hepg2亚克隆。‘kda’=千道尔顿(分子量标记)。图15d示出了使用抗idua抗体的蛋白质印迹(上图示出了暗曝光并且下图示出了亮曝光)以研究与可商购hidua(
图16a至16d描绘了添加的甘露糖6-磷酸酶(m6p)对hepg2细胞(图16a)或k562细胞(图16b)摄取ids的影响以及由k562细胞产生的idua摄取的图(图16c)。图16d是示出用甘露糖-6-磷酸处理靶细胞对摄取的影响的示意图。所描绘的数据说明了用m6p处理或保持未处理的细胞。在图16a和16b中,每个三元组,所示的样品,从左到右如下:最左边的条-包含用来自未处理的hepg2细胞的条件培养基处理的对照;中间条-用来自hepg2-ids克隆8的条件培养基处理的细胞;右边条-用来自hepg2-ids克隆15的条件培养基处理的细胞。在每个时间点处,指示用m6p处理的细胞。在指定的时间段后,使细胞沉淀并且然后测定ids活性。两种条件的比较说明了m6p抑制糖基化ids蛋白的摄取。图16c描绘了在从未处理的hepg2细胞或具有idua供体的hepg2细胞(idua克隆号25)获得的条件培养基中培养的k562细胞对idua的摄取。图16d是示出用甘露糖-6-磷酸(m6p)处理细胞的效果的示意图,说明了图16a-16c中的m6p处理如何阻断酶摄取,从而减少细胞中可检测的idua的量。
图17a和17b描绘了治疗和未治疗的野生型和mpsi和ii小鼠的脑中硫酸皮肤素(左图)和硫酸乙酰肝素(右图)水平的分析。图17a对应于图3b(ii)中分析的最左边的一组柱的小鼠。图17b对应于图9b中分析的最右边的一组柱的小鼠。
具体实施方式
本文公开了用于治疗和/或预防mpsi或mpsii疾病的方法和组合物。本发明提供了用于插入编码在患有mpsi或mpsii疾病的受试者中缺少或不充分表达的蛋白质的基因,使得所述基因在受试者的一种或多种细胞和/或组织中体内表达的方法和组合物。例如,所述基因可在靶组织中(例如,在肝脏中)表达以及从其在继代细胞和组织(例如,脾脏、肺、心脏、肌肉、脑等)中起作用的靶组织分泌。本发明还描述了细胞(例如,前体或成熟rbc、ipsc或肝细胞)的改变,使得其产生高水平的治疗剂,并且将这些改变的细胞群引入患者体内将提供所需的蛋白质。转基因可编码希望的蛋白质或结构rna,其在有需要的患者中是治疗学有益的。
因此,本发明的方法和组合物可用于表达来自任何基因座(例如,高度表达的白蛋白基因座)的转基因治疗学有益的mpsi和/或mpsii蛋白,以替换mpsi或mpsii疾病中缺陷的酶。另外,本发明提供了通过将转基因序列插入细胞诸如肝细胞中高度表达的基因座中来治疗mpsi和/或mpsii疾病(包括减轻一种或多种症状)的方法和组合物。
另外,可将转基因引入源自患者的细胞,例如,源自患者的诱导多能干细胞(ipsc)或其它类型的干细胞(胚胎或造血细胞)中用于最终植入。特别有用的是将治疗性转基因插入造血干细胞中以便植入有需要的患者体内。当干细胞分化成成熟细胞时,它们将含有高水平的治疗性蛋白以递送至组织。
综述
除非另外指示,否则这些方法的实施以及本文公开的组合物的制备和用途采用分子生物学、生物化学、染色质结构和分析、计算化学、细胞培养、重组dna以及本领域技术内的相关领域中的常规技术。这些技术在文献中被充分说明。参见例如,sambrook等molecularcloning:alaboratorymanual,第二版,coldspringharborlaboratorypress,1989以及第三版,2001;ausubel等,currentprotocolsinmolecularbiology,johnwiley&sons,newyork,1987以及定期更新;methodsinenzymology丛书,academicpress,sandiego;wolffe,chromatinstructureandfunction,第三版,academicpress,sandiego,1998;methodsinenzymology,第304卷,“chromatin”(p.m.wassarman以及a.p.wolffe编),academicpress,sandiego,1999;以及methodsinmolecularbiology,第119卷,“chromatinprotocols”(p.b.becker编)humanapress,totowa,1999。
定义
术语“核酸”、“多核苷酸”以及“寡核苷酸”可交换使用,并且是指呈直链或环状构象,以及呈单链或双链形式的脱氧核糖核苷酸或核糖核苷酸聚合物。出于本公开的目的,这些术语不应被解释为相对于聚合物的长度为限制性的。所述术语可涵盖天然核苷酸的已知类似物,以及在碱基、糖和/或磷酸部分(例如,硫代磷酸主链)中被修饰的核苷酸。通常地,特定核苷酸的类似物具有相同的碱基成对特异性;即a的类似物将会与t碱基成对。
术语“多肽”、“肽”以及“蛋白质”可交换使用,是指氨基酸残基的聚合物。所述术语还适用于其中一种或多种氨基酸是对应的天然存在氨基酸的化学类似物或修饰衍生物的氨基酸聚合物。
“结合”是指大分子之间(例如,蛋白质与核酸之间)序列特异、非共价相互作用。不是所有的结合相互作用组分都需要序列特异(例如,与dna主链中磷酸盐残基接触),只要相互作用总体上是序列特异即可。此类相互作用通常特征在于10-6m-1或更低的解离常数(kd)。“亲和力”是指结合的强度:增加的结合亲和力与较低kd有关。
“结合蛋白”是能够非共价结合另一分子的蛋白质。结合蛋白可结合至例如dna分子(dna结合蛋白)、rna分子(rna结合蛋白)和/或蛋白质分子(蛋白质结合蛋白)。就蛋白质结合蛋白来说,它可结合至其自身(以形成同源二聚体、同源三聚体等)和/或它可结合至不同蛋白质的一个或多个分子。结合蛋白可具有多于一种类型的结合活性。例如,锌指蛋白具有dna结合、rna结合以及蛋白质结合活性。
“锌指dna结合蛋白”(或结合结构域)是通过一个或多个锌指以序列特异方式结合dna的蛋白质,或较大蛋白质内的结构域,所述一个或多个锌指是其结构通过锌离子配位作用来稳定的结合结构域内的氨基酸序列区。术语锌指dna结合蛋白经常缩写为锌指蛋白或zfp。
“taledna结合结构域”或“tale”是包含一个或多个tale重复结构域/单元的多肽。重复结构域参与tale与其同源靶dna序列的结合。单个“重复单元”(也称为“重复”)的长度通常为33-35个氨基酸,并且与天然存在的tale蛋白中的其它tale重复序列表现出至少一些序列同源性。参见,例如美国专利号8,586,526。
锌指和tale结合结构域可被“工程化”以结合至预先确定的核苷酸序列,例如通过工程化(改变一个或多个氨基酸)天然存在的锌指或tale蛋白的识别螺旋区。因此,工程化dna结合蛋白(锌指或tale)是非天然存在的蛋白质。用于工程化dna结合蛋白的方法的非限制性实例是设计和选择。设计的dna结合蛋白是不会出现在自然中的蛋白质,所述蛋白质的设计/组成主要是合理标准的结果。用于设计的合理标准包括取代规则以及用于处理数据库中信息的计算算法的应用,所述数据库存储了现有zfp和/或tale设计和结合数据的信息。参见例如,美国专利号8,568,526;6,140,081;6,453,242;和6,534,261;还参见wo98/53058;wo98/53059;wo98/53060;wo02/016536和wo03/016496。
“选择的”锌指蛋白或tale是没有在自然中发现的蛋白质,所述蛋白质的生产主要是经验过程的结果,诸如噬菌体展示、相互捕获或杂种选择。参见例如,美国专利号8,586,526;5,789,538;5,925,523;6,007,988;6,013,453;6,200,759;以及wo95/19431;wo96/06166;wo98/53057;wo98/54311;wo00/27878;wo01/60970;wo01/88197和wo02/099084。
“重组”是指两个多核苷酸之间基因信息交换的过程。出于本公开的目的,“同源重组(hr)”是指这种交换的特定形式,所述交换发生在例如通过同源导向修复机制所进行的细胞中双链断裂的修复期间。此过程需要核苷酸序列同源性,使用“供体”分子以模板修复“靶”分子(即,经历双链断裂的分子),并且此过程被不同地称为“非交换型基因转化”或“短序列基因转化(shorttractgeneconversion),因为它导致基因信息从供体向靶转移。不希望被任何具体理论束缚,这种转移可涉及在断裂的靶标与供体之间形成的异源双链dna的错配校正,和/或“合成依赖性链退火”,其中供体用于再合成将成为靶标的一部分的遗传信息;和/或相关过程。这种特定的hr经常导致靶分子序列的改变,使得供体多核苷酸的部分或所有序列并入靶多核苷酸中。
在本公开的方法中,如本文所述的一种或多种靶向核酸酶可在预先确定的位点处,在靶序列(例如,细胞染色质)中产生双链断裂,并且与断裂区中的核苷酸序列具有同源性的“供体”多核苷酸可被引入细胞中。双链断裂的存在已被证明有利于供体序列的整合。供体序列可被物理整合或者可替代地,供体多核苷酸被用作用于通过同源组合来修复断裂的模板,从而导致将供体中的全部或部分核苷酸序列引入细胞染色质中。因此,可改变细胞染色质中的第一序列,并且在某些实施方案中,可将其转化成存在于供体多核苷酸中的序列。因此,术语“替换”或“替代”的使用可理解为表示用另一核苷酸序列替代一个核苷酸序列,(即,在信息意义上替代序列),并且不一定需要用另一多核苷酸物理或化学地替代一个多核苷酸。
在本文所述的任何方法中,另外的锌指对或talen蛋白可用于细胞内另外靶位点的另外双链裂解。
在用于靶向重组和/或替代和/或改变细胞染色质中感兴趣区域序列的方法的实施方案中,染色体序列通过用外源“供体”核苷酸序列同源重组来改变。如果存在与断裂区同源的序列,则通过细胞染色质中存在双链断裂来刺激这种同源重组。
在本文所述的任何方法中,第一核苷酸序列(“供体序列”)可含有与感兴趣区域中的基因组序列同源但不同的序列,从而刺激同源重组以在感兴趣区域中插入不相同的序列。因此,在某些实施方案中,与感兴趣区域中的序列同源的供体序列部分表现出与被替换的基因组序列约80%至99%之间(或其间的任何整数)的序列同一性。在其它实施方案中,供体与基因组序列之间的同源性高于99%,例如如果具有超过100个邻接碱基对的供体与基因组序列之间仅有1个核苷酸不同。在某些情况下,供体序列的非同源部分可含有不存在于感兴趣区域中的序列,使得新的序列引入感兴趣区域中。在这些情况下,非同源序列通常侧接具有50-1,000个碱基对(或其间的任何整数值)或大于1,000的任何数量碱基对的序列,这些序列与感兴趣区域中的序列同源或相同。在其它实施方案中,供体序列与第一序列是非同源的,并且通过非同源重组机制插入基因组中。
本文所述的任何方法可用于通过靶向整合破坏感兴趣基因表达的供体序列来使细胞中的一种或多种靶序列部分或完全失活。还提供了具有部分或完全失活基因的细胞系。
此外,如本文所述的靶向整合方法还可用于整合一种或多种外源序列。外源核酸序列可包含,例如一种或多种基因或cdna分子、或任何类型的编码或非编码的序列,以及一种或多种控制元件(例如,启动子)。另外,外源核酸序列可产生一种或多种rna分子(例如,小发夹rna(shrna)、抑制性rna(rnai)、微rna(mirna)等)。
“裂解”是指dna分子共价主链的断裂。裂解可通过多种方法来引发,包括但不限于磷酸二酯键的酶水解或化学水解。单链裂解和双链裂解两者都是可能的,并且双链裂解可由于两个不同单链裂解事件而发生。dna裂解可导致平端或交错端的产生。在某些实施方案中,融合多肽用于靶向双链dna裂解。
“裂解半结构域”是与第二多肽(相同的或不同的)连结,形成具有裂解活性(优选地双链裂解活性)的复合物的多肽序列。术语“第一和第二裂解半结构域”;“+和-裂解半结构域”以及“右和左裂解半结构域”可交换使用,是指二聚化的裂解半结构域对。
“工程化裂解半结构域”是已被修饰以便与另一裂解半结构域(例如,另一工程化裂解半结构域)形成专性异二聚体的裂解半结构域。参见美国专利号7,888,121;7,914,796;8,034,598和8,823,618,其以引用方式全文并入本文。
术语“序列”是指任何长度的核苷酸序列,其可以是dna或rna;可以是直链的、环状的或支链的,并且可以是单链或双链的。术语“供体序列”是指插入基因组中的核苷酸序列。供体序列可以是任何长度,例如2与10,000个核苷酸之间的长度(或其间或其上的任何整数值),优选地约100与1,000个核苷酸之间的长度(或其间的任何整数值),更优选地约200与500个核苷酸之间的长度。
“疾病相关基因”是在单基因疾病中以某种方式缺陷的基因。单基因疾病的非限制性实例包括严重联合免疫缺陷、囊性纤维化、溶酶体贮积病(例如gaucher病、hurler病、hunter病、fabry病、neimann-pick病、tay-sach病等)、镰状细胞性贫血和地中海贫血。
“血脑屏障”是一种高度选择性渗透屏障,它将循环血液与中枢神经系统中的脑分开。血脑屏障由脑内皮细胞形成,所述脑内皮细胞通过cns血管中限制血液溶质通过的紧密连接而连接。长期以来一直认为血脑屏障阻止大分子治疗剂的摄取并阻止大多数小分子治疗剂的摄取(pardridge(2005)neurorx2(1):3-14)。
“染色质”是包含细胞基因组的核蛋白结构。细胞染色质包含核酸(主要为dna)和蛋白质,包括组蛋白和非组蛋白的染色体蛋白。大多数真核细胞染色质以核小体的形式存在,其中核小体核心包含大约150个与八聚物相关的dna碱基对,所述八聚物各自包含组蛋白h2a、h2b、h3和h4中的两个;并且接头dna(取决于有机体具有可变长度)在核小体核心之间延伸。组蛋白h1的分子一般与接头dna相关。出于本公开的目的,术语“染色质”意在包括所有类型的细胞核蛋白,不论是原核的还是真核的。细胞染色质包括染色体染色质和游离基因染色质两者。
“染色体”是包含细胞全部或部分基因组的染色质复合物。细胞的基因组经常以其核型为特征,所述核型是构成细胞基因组的所有染色体的集合。细胞的基因组可包含一个或多个染色体。
“游离基因”是复制的核酸、核蛋白复合物或包含不是细胞染色体核型的一部分的核酸的其它结构。游离基因的实例包括质粒和某些病毒基因组。
“靶位点”或“靶序列”是限定结合分子将结合(只要存在足够的结合条件)的核酸的一部分的核酸序列。
“外源”分子是通常不存在于细胞中,但可通过一种或多种基因的、生物化学的或其它方法引入细胞中的分子。“正常存在于细胞中”是相对于细胞的特定发育阶段和环境条件确定的。因此,例如,仅在肌肉的胚胎发育期间存在的分子是相对于成体肌肉细胞的外源分子。类似地,由热休克诱导的分子是相对于非热休克细胞的外源分子。外源分子可包含,例如功能失常的内源分子的功能型式或功能正常的内源分子的功能失常型式。
除其它外,外源分子可以是小分子,诸如通过组合化学过程产生的小分子,或大分子诸如蛋白质、核酸、碳水化合物、脂质、糖蛋白、脂蛋白、多糖、上述分子的任何修饰衍生物或包含一种或多种上述分子的任何复合物。核酸包括dna和rna,其可以是单链或双链的;可以是直链、支链或环状的;并且可以是任何长度。核酸包括能够形成双链体以及三链体的那些核酸。参见例如,美国专利号5,176,996和5,422,251。蛋白质包括但不限于dna结合蛋白、转录因子、染色质重塑因子、甲基化dna结合蛋白、聚合酶、甲基化酶、脱甲基酶、乙酰基转移酶、脱乙酰酶、激酶、磷酸酶、整合酶、重组酶、连接酶、拓扑异构酶、回旋酶以及解旋酶。
外源分子可以是与内源分子相同类型的分子,例如外源蛋白质或核酸。例如,外源核酸可包含感染的病毒基因组、引入细胞中的质粒或游离基因、或通常不存在于细胞中的染色体。用于将外源分子引入细胞中的方法是本领域技术人员已知的,并且包括但不限于脂质介导的转移(即,脂质体,包括中性和阳离子脂质)、电穿孔、直接注入、细胞融合、粒子轰击、磷酸钙共沉淀、deae-葡聚糖介导的转移以及病毒载体介导的转移。外源分子还可以是与内源分子相同类型的分子,但源自与细胞源自的不同物种。例如,可将人核酸序列引入最初源自小鼠或仓鼠的细胞系中。
相反,“内源”分子是在特定环境条件下,在特定发育阶段通常存在于特定细胞中的分子。例如,内源核酸可包含染色体、线粒体、叶绿体或其它细胞器的基因组,或天然存在的游离核酸。另外的内源分子可包括蛋白质,例如转录因子和酶。
“融合”分子是其中两个或更多个亚单位分子优选地共价连接的分子。亚单位分子可以是相同化学类型的分子,或可以是不同化学类型的分子。融合分子的实例包括但不限于融合蛋白(例如,蛋白质dna结合结构域与裂解结构域之间的融合)、与裂解结构域操作性地关联的多核苷酸dna结合结构域之间的融合体(例如,sgrna),以及融合核酸(例如,编码融合蛋白的核酸)。
融合蛋白在细胞中的表达可通过将融合蛋白递送至细胞或通过将编码融合蛋白的多核苷酸递送至细胞引起,其中多核苷酸被转录,并且转录物被转录,以产生融合蛋白。反式剪接、多肽裂解和多肽连接还可参与细胞中的蛋白质表达。用于将多核苷酸和多肽递送至细胞的方法在本公开的其它地方提出。
出于本公开的目的,“基因”包括编码基因产物的dna区(参见下文),以及调节基因产物产生的所有dna区,无论此类调节序列是否与编码和/或转录序列邻近。因此,基因包括但不一定限于启动子序列、终止子、翻译调节序列诸如核糖体结合位点和内部核糖体进入位点、增强子、沉默子、绝缘子、边界元件、复制起点、基质附着位点以及基因座控制区。
“基因表达”是指将包含在基因中的信息转化成基因产物。基因产物可以是基因的直接转录产物(例如,mrna、trna、rrna、反义rna、核酶、结构rna或任何其它类型的rna)或通过mrna翻译产生的蛋白质。基因产物还包括通过诸如加帽化、聚腺苷酸化、甲基化以及编辑的方法修饰的rna,以及通过例如甲基化、乙酰化、磷酸化、遍在蛋白化、adp-核糖基化、肉豆蔻基化以及糖基化修饰的蛋白质。
基因表达的“调节”是指基因活性的变化。表达的调节可包括但不限于基因激活和基因阻抑。基因组编辑(例如,裂解、改变、失活、随机突变)可用于调节表达。基因失活是指与不包括如本文所述的zfp或talen的细胞相比,基因表达的任何减少。因此,基因失活可能是部分的或完全的。
“感兴趣区域”是细胞染色质的任何区域,例如像基因或者基因内或附近的非编码序列,其中结合外源分子是希望的。结合可以是出于靶向dna裂解和/或靶向重组的目的。感兴趣区域可存在于例如染色体、游离基因、细胞器基因组(例如,线粒体、叶绿体)或感染病毒基因组中。感兴趣区域可在基因的编码区内,在转录的非编码区内,例如像前导序列、尾随序列或内含子,或在编码区上游或下游的非转录区内。感兴趣区域可小至单个核苷酸对,或高达2,000个核苷酸对长度,或任何整数值的核苷酸对。
“真核”细胞包括但不限于真菌细胞(例如酵母)、植物细胞、动物细胞、哺乳动物细胞和人细胞(例如,t细胞)。
“红细胞”(rbc)或红血球是源自造血干细胞的终末分化细胞。它们缺少核酸酶和大多数细胞器。rbc含有用于将氧气从肺输送至外周组织的血红蛋白。事实上,33%的个体rbc是血红蛋白。它们还在代谢过程中将由细胞产生的co2从组织中带出并返回至肺部,以便在呼气时释放。rbc在骨髓中响应于血液缺氧而产生,其是由肾脏释放促红细胞生成素(epo)介导的。epo引起原红细胞数量的增加并缩短完全rbc成熟所需的时间。在大约120天后,由于rbc不含有细胞核或任何其它再生能力,因此通过肝脏、脾脏和淋巴结中巨噬细胞的吞噬活性(约90%)或通过血浆中的溶血(约10%)将细胞从循环中去除。在巨噬细胞吞噬后,由于溶酶体酶的作用,rbc的化学成分在巨噬细胞的空泡中被分解。
“分泌组织”是动物中将个体细胞的产物分泌到某些类型腔中的那些组织,其通常源自上皮细胞。定位于胃肠道的分泌组织的实例包括排列肠、胰腺和胆囊的细胞。其它分泌组织包括肝脏、与眼睛相关的组织和粘膜诸如唾液腺、乳腺、前列腺、脑垂体和内分泌系统的其它成员。另外,分泌组织包括能够分泌的组织类型的单个细胞。
术语“操作性连接”和“操作性地连接”(或“可操作地连接”)关于并置两种或更多种组分(诸如序列元件)可交换使用,其中组分被布置成使得两种组分都功能正常并且允许组分中的至少一种可介导施加在其它组分中的至少一种上的功能。作为说明,如果转录调节序列响应于一种或多种转录调节因子的存在或不存在而控制编码序列的转录水平,则转录调节序列诸如启动子操作性地连接至编码序列。转录调节序列通常以顺式与编码序列操作性地连接,但不需要与其直接相邻。例如,增强子是与编码序列操作性地连接的转录调节序列,即使它们不是邻接的。
关于融合多肽,术语“操作性地连接”可以是指以下事实:每个组分在与其它组分的连接状态下执行与其在未这样连接的情况下所执行的功能相同的功能。例如,关于其中zfp或taledna结合结构域融合至激活结构域的融合多肽,如果在融合多肽中,zfp或taledna结合结构域部分能够结合其靶位点和/或其结合位点,同时激活结构域能够上调基因表达,则zfp或taledna结合结构域和激活结构域是操作性连接的。当为其中zfp或taledna结合结构域融合至裂解结构域的融合多肽时,如果在融合多肽中,zfp或taledna结合结构域部分能够结合其靶位点和/或其结合位点,同时裂解结构域能够裂解靶位点附近的dna,则zfp或taledna结合结构域和裂解结构域是操作性连接的。
蛋白质、多肽或核酸的“功能片段”是其序列不同于全长蛋白质、多肽或核酸,但仍保留与全长蛋白质、多肽或核酸相同功能的蛋白质、多肽或核酸。功能片段可具有比对应天然分子更多、更少或相同数量的残基,和/或可含有一种或多种氨基酸或核苷酸取代。用于测定核酸功能(例如,编码功能,与另一核酸杂交的能力)的方法是本领域众所周知的。类似地,用于测定蛋白质功能的方法是众所周知的。例如,多肽的dna结合功能可例如通过过滤结合、电泳迁移率改变或免疫沉淀测定法来测定。dna裂解可通过凝胶电泳来测定。参见ausubel等,同上。蛋白质与另一蛋白质相互作用的能力可通过例如共免疫沉淀、双杂交测定或基因和生物化学的两种互补作用来测定。参见例如,fields等(1989)nature340:245-246;美国专利号5,585,245和pctwo98/44350。
“载体”能够将基因序列转移至靶细胞。通常,“载体构建体”、“表达载体”和“基因转移载体”意指能够指导感兴趣基因的表达并且可将基因序列转移至靶细胞的任何核酸构建体。因此,所述术语包括克隆和表达载体,以及整合载体。
“报告基因”或“报告序列”是指产生易于测量的蛋白质产物的任何序列,优选但不一定在常规测定中。合适的报告基因包括但不限于编码介导抗生素抗性(例如,氨苄青霉素抗性、新霉素抗性、g418抗性、嘌呤霉素抗性)的蛋白质的序列、编码有色或荧光或发光蛋白(例如,绿色荧光蛋白、增强的绿色荧光蛋白、红色荧光蛋白、荧光素酶)和介导增强的细胞生长和/或基因扩增的蛋白质(例如,二氢叶酸还原酶)的序列。表位标签包括,例如,flag、his、myc,tap、ha或任何可检测的氨基酸序列中的一个或多个拷贝。“表达标签”包括编码报告子的序列,所述报告子可操作地连接至希望的基因序列,以便监测感兴趣基因的表达。
术语“受试者”或“患者”可交换使用并且是指哺乳动物诸如人患者和非人灵长类动物以及实验动物诸如兔、狗、猫、大鼠、小鼠以及其它动物。因此,如本文所用的术语“受试者”或“患者”意指可向其施用本发明的改变的细胞和/或由本发明的改变的细胞产生的蛋白质的任何哺乳动物患者或受试者。本发明的受试者包括具有lsd(mpsi)和/或mpsii病症的那些受试者。
核酸酶
本文所述的方法可利用一种或多种核酸酶以便靶向引入idua或ids转基因。核酸酶的非限制性实例包括zfn、talen、归巢内切核酸酶、crispr/cas和/或ttago指导rna,其可用于体内裂解携带转基因和核酸酶的供体分子以便裂解细胞的基因组,使得转基因以靶向的方式整合到基因组中。在某些实施方案中,核酸酶中的一种或多种是天然存在的。在其它实施方案中,核酸酶中的一种或多种是非天然存在的,即在dna结合分子(也称为dna结合结构域)和/或裂解结构域中进行工程化。例如,可改变天然存在的核酸酶的dna结合结构域以结合至选定靶位点(例如,被工程化以结合至选定靶位点的zfp、tale和/或crispr/cas的sgrna)。在其它实施方案中,核酸酶包含异源dna结合结构域和裂解结构域(例如,锌指核酸酶;tal-效应子结构域dna结合蛋白;具有异源裂解结构域的大范围核酸酶dna结合结构域)。在其它实施方案中,核酸酶包含系统,诸如ttago系统的crispr/cas。
a.dna结合结构域
在某些实施方案中,本文所述的组合物和方法采用大范围核酸酶(归巢内切核酸酶)dna结合结构域用于结合至供体分子和/或结合至细胞基因组中的感兴趣区域。天然存在的大范围核酸酶识别15-40个碱基对裂解位点,并且通常分为四个家族:laglidadg家族、giy-yig家族、his-cyst盒家族和hnh家族。示例性的归巢内切核酸酶包括i-scei、i-ceui、pi-pspi、pi-sce、i-sceiv、i-csmi、i-pani、i-sceii、i-ppoi、i-sceiii、i-crei、i-tevi、i-tevii和i-teviii。它们的识别序列是已知的。还参见美国专利号5,420,032;美国专利号6,833,252;belfort等(1997)nucleicacidsres.25:3379–3388;dujon等(1989)gene82:115–118;perler等(1994)nucleicacidsres.22,1125–1127;jasin(1996)trendsgenet.12:224–228;gimble等(1996)j.mol.biol.263:163–180;argast等(1998)j.mol.biol.280:345–353以及新英格兰生物实验室目录(thenewenglandbiolabscatalogue)。
在某些实施方案中,本文所述的方法和组合物利用包含工程化(非天然存在的)归巢内切核酸酶(大范围核酸酶)的核酸酶。归巢内切核酸酶和大范围核酸酶的识别序列,诸如i-scei、i-ceui、pi-pspi、pi-sce、i-sceiv、i-csmi、i-pani、i-sceii、i-ppoi、i-sceiii、i-crei、i-tevi、i-tevii和i-teviiii都是已知的。还参见美国专利号5,420,032;美国专利号6,833,252;belfort等(1997)nucleicacidsres.25:3379–3388;dujon等(1989)gene82:115–118;perler等(1994)nucleicacidsres.22,1125–1127;jasin(1996)trendsgenet.12:224–228;gimble等(1996)j.mol.biol.263:163–180;argast等(1998)j.mol.biol.280:345–353以及新英格兰生物实验室目录。另外,可工程化归巢内切核酸酶和大范围核酸酶的dna结合特异性以结合非天然的靶位点。参见例如,chevalier等(2002)molec.cell10:895-905;epinat等(2003)nucleicacidsres.31:2952-2962;ashworth等(2006)nature441:656-659;paques等(2007)currentgenetherapy7:49-66;美国专利公布号20070117128。归巢内切核酸酶和大范围核酸酶的dna结合结构域可在整体的核酸酶背景下改变(即,使得核酸酶包括同源裂解结构域)或可融合至异源裂解结构域。
在其它实施方案中,本文所述的方法和组合物中使用的一种或多种核酸酶的dna结合结构域包含天然存在的或工程化的(非天然存在的)tal效应子dna结合结构域。参见例如,美国专利号8,586,526,其以引用方式全文并入本文。已知黄单胞菌属的植物病原菌在重要的作物植物中引起许多疾病。黄单胞菌的病原性取决于保守的iii型分泌(t3s)系统,其将超过25种不同的效应蛋白注入植物细胞中。在这些注射的蛋白质中的是转录激活因子样(tal)效应子,其模拟植物转录激活因子并操纵植物转录组(参见kay等(2007)science318:648-651)。这些蛋白质含有dna结合结构域和转录激活结构域。最充分表征的tal效应子之一是来自xanthomonascampestgrispv.vesicatoria的avrbs3(参见bonas等(1989)molgengenet218:127-136和wo2010079430)。tal效应子含有串联重复序列的集中结构域,每个重复含有大约34个氨基酸,这对于这些蛋白质的dna结合特异性是关键的。另外,它们含有核定位序列和酸性转录激活结构域(综述参见schornacks等(2006)jplantphysiol163(3):256-272)。另外,在植物致病细菌ralstoniasolanacearum中,已发现两个基因,命名为brg11和hpx17,与r.solanacearumbiovar1菌株gmi1000和生物变种4菌株rs1000中的xanthomonas的avrbs3家族同源(参见heuer等(2007)applandenvirmicro73(13):4379-4384)。这些基因在核苷酸序列上彼此98.9%相同,但在hpx17的重复结构域中相差1,575bp的缺失。然而,两种基因产物与xanthomonas的avrbs3家族蛋白具有小于40%的序列同一性。参见例如,美国专利号8,586,526,其以引用方式全文并入本文。
这些tal效应子的特异性取决于在串联重复序列中发现的序列。重复序列包含大约102bp,并且重复序列通常彼此91%-100%同源(bonas等,同上)。重复序列的多态性通常位于位置12和13,并且在位置12和13的高变双残基(rvd)的同一性与tal效应子的靶序列中连续核苷酸的同一性之间似乎存在一对一的对应关系(参见moscou和bogdanove,(2009)science326:1501和boch等(2009)science326:1509-1512)。实验上,已确定了这些tal效应子的dna识别的天然代码,使得位置12和13的hd序列导致与胞嘧啶(c)的结合,ng与t结合,ni与a、c、g或t结合,nn与a或g结合以及ing与t结合。这些dna结合重复序列已组装成具有新组合和重复数量的蛋白质,以制造能够与新序列相互作用并激活植物细胞中非内源报告基因表达的人工转录因子(boch等,同上)。已将工程化tal蛋白与foki裂解半结构域连接,以产生在酵母报告基因测定(基于质粒的靶标)中表现出活性的tal效应子结构域核酸酶融合体(talen)。参见例如,美国专利号8,586,526;christian等((2010)geneticsepub10.1534/genetics.110.120717)。
在某些实施方案中,用于体内裂解和/或靶向裂解细胞基因组的一种或多种核酸酶的dna结合结构域包含锌指蛋白。优选地,锌指蛋白是非天然存在的,因为其被工程化以结合至选择的靶位点。参见例如,beerli等(2002)naturebiotechnol.20:135-141;pabo等(2001)ann.rev.biochem.70:313-340;isalan等(2001)naturebiotechnol.19:656-660;segal等(2001)curr.opin.biotechnol.12:632-637;choo等(2000)curr.opin.struct.biol.10:411-416;美国专利号6,453,242;6,534,261;6,599,692;6,503,717;6,689,558;7,030,215;6,794,136;7,067,317;7,262,054;7,070,934;7,361,635;7,253,273;以及美国专利公布号2005/0064474;2007/0218528;2005/0267061,所有文献均以引用方式全文并入本文。
与天然存在的锌指蛋白相比,工程化的锌指结合结构域可具有新颖的结合特异性。工程化方法包括但不限于合理设计和各种类型的选择。合理设计包括,例如,使用包含三联体(或四联体)核苷酸序列和单个锌指氨基酸序列的数据库,其中每个三联体或四联体核苷酸序列与锌指的一个或多个氨基酸序列相关,所述锌指结合特定的三联体或四联体序列。参见例如,共有的美国专利6,453,242和6,534,261,其以引用方式全文并入本文。
包括噬菌体展示和双杂交系统的示例性选择方法公开于美国专利5,789,538;5,925,523;6,007,988;6,013,453;6,410,248;6,140,466;6,200,759;和6,242,568;以及wo98/37186;wo98/53057;wo00/27878;和wo01/88197。另外,已在例如共有的wo02/077227中描述了用于锌指结合结构域的结合特异性增强。
另外,如在这些和其它参考文献中所公开的,锌指结构域和/或多指锌指蛋白可使用任何合适的接头序列连接在一起,包括例如长度为五个或更多个氨基酸的接头。关于示例性接头序列,还参见美国专利号8,772,453;6,479,626;6,903,185;和7,153,949。本文所述的蛋白质可包括蛋白质的单个锌指之间的合适接头的任何组合。
靶位点的选择;zfp和用于设计和构建融合蛋白(和编码其的多核苷酸)的方法是本领域技术人员已知的并且详细描述于美国专利号6,140,081;5,789,538;6,453,242;6,534,261;5,925,523;6,007,988;6,013,453;6,200,759;wo95/19431;wo96/06166;wo98/53057;wo98/54311;wo00/27878;wo01/60970;wo01/88197;wo02/099084;wo98/53058;wo98/53059;wo98/53060;wo02/016536和wo03/016496中。
另外,如在这些和其它参考文献中所公开的,锌指结构域和/或多指锌指蛋白可使用任何合适的接头序列连接在一起,包括例如长度为五个或更多个氨基酸的接头。关于示例性6个或更多个氨基酸长度的接头序列,还参见美国专利号6,479,626;6,903,185;和7,153,949。本文所述的蛋白质可包括蛋白质的单个锌指之间的合适接头的任何组合。
在某些实施方案中,核酸酶的dna结合结构域是crispr/cas核酸酶系统的一部分,包括例如单指导rna(sgrna)。参见例如,美国专利号8,697,359和美国专利公布号20150056705。编码系统的rna组分的crispr(规律间隔的短回文重复序列)基因座,和编码蛋白质的cas(crispr相关)基因座(jansen等,2002.mol.microbiol.43:1565-1575;makarova等,2002.nucleicacidsres.30:482-496;makarova等,2006.biol.direct1:7;haft等,2005.ploscomput.biol.1:e60)组成crispr/cas核酸酶系统的基因序列。微生物宿主中的crispr基因座含有crispr相关(cas)基因的组合以及能够编程crispr介导的核酸裂解的特异性的非编码rna元件。
ii型crispr是最充分表征的系统之一,并在四个连续步骤中进行靶向dna双链断裂。首先,两个非编码rna、前体crrna(pre-crrna)阵列和反式激活的crrna(tracrrna),从crispr基因座转录。然后,反式激活的crrna与前体crrna的重复区域杂交并介导前体crrna加工成含有单个间隔区序列的成熟crrna。第三,成熟的crrna:反式激活的crrna复合物通过crrna上的间隔区与靶dna上的原型间隔区之间的watson-crick碱基配对将cas9引导至靶dna,所述原型间隔区的旁边是原型间隔区相邻基序(pam),其是靶识别的另一个要求。最后,cas9介导靶dna的裂解以在原型间隔区内产生双链断裂。crispr/cas系统的活动包括三个步骤:(i)在称为‘适应’的过程中,将外来dna序列插入crispr阵列中以防止未来的攻击,(ii)相关蛋白的表达,以及阵列的表达和加工,随后是(iii)rna介导的干扰外来核酸。因此,在细菌细胞中,几种所谓的‘cas’蛋白与crispr/cas系统的天然功能有关,并在诸如插入外来dna等功能中起作用。
在某些实施方案中,cas蛋白可以是天然存在的cas蛋白的“功能衍生物”。天然序列多肽的“功能衍生物”是具有与天然序列多肽共同的定性生物学特性的化合物。“功能衍生物”包括但不限于天然序列的片段和天然序列多肽及其片段的衍生物,条件是它们具有与相应的天然序列多肽共有的生物学活性。本文考虑的生物活性是功能衍生物将dna底物水解成片段的能力。术语“衍生物”包括多肽的氨基酸序列变体、共价修饰及其融合体。合适的cas多肽或其片段的衍生物包括但不限于cas蛋白的突变体、融合体、共价修饰或其片段。cas蛋白(包括cas蛋白或其片段),以及cas蛋白或其片段的衍生物,可从细胞中获得或化学合成或通过这两种程序的组合获得。细胞可以是天然产生cas蛋白的细胞,或天然产生cas蛋白并且被基因工程化以更高的表达水平产生内源cas蛋白或由外源引入的核酸产生cas蛋白的细胞,所述核酸编码与内源cas相同或不同的cas。在一些情况下,细胞不天然产生cas蛋白,并且被基因工程化以产生cas蛋白。可与cas蛋白一起使用和/或代替cas蛋白使用的rna引导核酸酶的另外非限制性实例包括2类crispr蛋白,诸如cpf1。参见例如,zetsche等(2015)cell163:1-13。
在一些实施方案中,dna结合结构域是ttago系统的一部分(参见swarts等,同上;sheng等,同上)。在真核生物中,基因沉默由argonaute(ago)蛋白家族介导。在这个范例中,ago与小(19-31nt)rna结合。此蛋白质-rna沉默复合物通过小rna与靶标之间的watson-crick碱基配对识别靶rna并且内切核酸裂解靶rna(vogel(2014)science344:972-973)。相比之下,原核生物ago蛋白与小单链dna片段结合,并且可能起到检测和去除外来(经常是病毒)dna的作用(yuan等,(2005)mol.cell19,405;olovnikov等(2013)mol.cell51,594;swarts等,同上)。示例性的原核生物ago蛋白包括来自aquifexaeolicus、类球红细菌(rhodobactersphaeroides)和嗜热栖热菌(thermusthermophilus)的那些。
最充分表征的原核生物ago蛋白之一来自嗜热链球菌(t.thermophilus)(ttago;swarts等,同上)。ttago与15nt或13-25nt的具有5'磷酸酯基团的单链dna片段相联。由ttago结合的这种“指导dna”用于指导蛋白质-dna复合物结合第三方dna分子中的watson-crick互补dna序列。一旦这些指导dna中的序列信息允许鉴定靶dna,则ttago-指导dna复合物裂解靶dna。这种机制还受到ttago-指导dna复合物的结构的支持,同时与其靶dna结合(g.sheng等,同上)。来自类球红细菌的ago(rsago)具有类似的特性(olivnikov等,同上)。
任意dna序列的外源指导dna可加载到ttago蛋白上(swarts等,同上)。由于ttago裂解的特异性是由指导dna指导的,因此用外源的、研究者指定的指导dna形成的ttago-dna复合物将指导ttago靶dna裂解成互补的研究者指定的靶dna。以这种方式,可在dna中产生靶向双链断裂。使用ttago-指导dna系统(或来自其它生物的直系同源ago-指导dna系统)允许靶向裂解细胞内的基因组dna。这种裂解可以是单链或双链的。对于哺乳动物基因组dna的裂解,优选使用经密码子优化以在哺乳动物细胞中表达的ttago型式。此外,可能优选用体外形成的ttago-dna复合物处理细胞,其中ttago蛋白融合至细胞穿透肽。此外,可能优选使用通过诱变而被改变的ttago蛋白的型式以在37摄氏度下具有改善的活性。ttago-rna介导的dna裂解可用于使用本领域针对利用dna断裂的标准技术实现多种结果,包括基因敲除、靶基因添加、基因校正、靶基因缺失。
因此,核酸酶包含dna结合结构域,其特异性结合至任何基因(其中希望插入供体(转基因))中的靶位点。
b.裂解结构域
任何合适的裂解结构域可与dna结合结构域相联(例如,操作性地连接)以形成核酸酶。例如,zfpdna结合结构域已融合至核酸酶结构域以产生zfn-一种能够通过其工程化(zfp)dna结合结构域识别其预期核酸靶标并通过核酸酶活性导致dna在zfp结合位点附近被切割的功能实体。参见例如,kim等(1996)procnatlacadsciusa93(3):1156-1160。最近,zfn已被用于各种生物中的基因组修饰。参见例如,美国专利公布20030232410;20050208489;20050026157;20050064474;20060188987;20060063231;和国际公布wo07/014275。同样,taledna结合结构域已融合至核酸酶结构域以产生talen。参见,例如美国专利号8,586,526。还描述了包含与dna结合并与裂解结构域(例如,cas结构域)相联以诱导靶向裂解的单指导rna(sgrna)的crispr/cas核酸酶系统。参见例如,美国专利号8,697,359和8,932,814以及美国专利公布号20150056705。
如上所述,裂解结构域可与dna结合结构域异源,例如锌指dna结合结构域和来自核酸酶的裂解结构域或talendna结合结构域和来自核酸酶的裂解结构域;sgrnadna结合结构域和来自核酸酶的裂解结构域(crispr/cas);和/或大范围核酸酶dna结合结构域和来自不同核酸酶的裂解结构域。异源裂解结构域可从任何内切核酸酶或外切核酸酶获得。可从其得到裂解结构域的示例性内切核酸酶包括但不限于限制性内切核酸酶和归巢内切核酸酶。参见例如,2002-2003catalogue,newenglandbiolabs,beverly,ma;和belfort等(1997)nucleicacidsres.25:3379-3388。裂解dna的另外的酶是已知的(例如,s1核酸酶;绿豆核酸酶;胰腺dna酶i;微球菌核酸酶;酵母ho内切核酸酶;还参见linn等(编)nucleases,coldspringharborlaboratorypress,1993)。这些酶(或其功能片段)中的一种或多种可用作裂解结构域和裂解半结构域的来源。
类似地,裂解半结构域可源自任何核酸酶或其部分,如上所阐述,其需要二聚作用以具有裂解活性。通常地,如果融合蛋白包含裂解半结构域,则裂解需要两个融合蛋白。可替代地,可使用包含两个裂解半结构域的单个蛋白质。两个裂解半结构域可源自相同的内切核酸酶(或其功能片段),或者每个裂解半结构域可源自不同的内切核酸酶(或其功能片段)。另外,两个融合蛋白的靶位点优选地相对于彼此布置,使得两个融合蛋白与它们各自的靶位点的结合将裂解半结构域放置于使得裂解半结构域例如通过二聚作用以形成功能裂解结构域的彼此空间定位中。因此,在某些实施方案中,靶位点的近边缘被5-8个核苷酸或被15-18个核苷酸隔开。然而,任何整数数量的核苷酸或核苷酸对可介于两个靶位点之间(例如,2至50个核苷酸对或更多)。通常地,裂解的位点位于靶位点之间。
限制性内切核酸酶(限制性酶)存在于许多物种中并且能够(在识别位点处)序列特异性地结合至dna,并在结合位点处或附近裂解dna。某些限制性酶(例如,iis型)在远离识别位点的位点处裂解dna并且具有可分隔的结合结构域和裂解结构域。例如,iis型酶foki在离一条链上识别位点的9个核苷酸处和在离另一条链上的识别位点13个核苷酸处催化dna的双链裂解。参见,例如美国专利5,356,802;5,436,150和5,487,994;以及li等(1992)proc.natl.acad.sci.usa89:4275-4279;li等(1993)proc.natl.acad.sci.usa90:2764-2768;kim等(1994a)proc.natl.acad.sci.usa91:883-887;kim等(1994b)j.biol.chem.269:31,978-31,982。因此,在一个实施方案中,融合蛋白包含来自至少一种iis型限制性酶的裂解结构域(或裂解半结构域)和一个或多个锌指结合结构域,所述锌指结合结构域可能是或可能不是工程化的。
其裂解结构域与结合结构域可分隔的示例性iis型限制性酶是foki。此特定酶作为二聚体具有活性。bitinaite等(1998)proc.natl.acad.sci.usa95:10,570-10,575。因此,出于本公开的目的,认为在所公开的融合蛋白中使用的foki酶部分是裂解半结构域。因此,对于使用锌指-foki融合体的靶向双链裂解和/或靶向替代细胞序列,可使用两个融合蛋白(各包含foki裂解半结构域)以重新构成催化活性的裂解结构域。可替代地,还可使用含有锌指结合结构域和两个foki裂解半结构域的单个多肽分子。使用锌指-foki融合体靶向裂解和靶向序列改变的参数在本公开的其它地方提供。
裂解结构域或裂解半结构域可以是蛋白质的任何部分,其保留裂解活性或保留多聚化(例如,二聚化)以形成功能性裂解结构域的能力。
示例性的iis型限制性酶描述于美国专利7,888,121中,其全文并入本文。另外的限制性酶也含有可分隔的结合结构域和裂解结构域,并且这些酶是本公开涵盖的。参见例如,roberts等(2003)nucleicacidsres.31:418-420。
在某些实施方案中,裂解结构域包含最小化或防止同源二聚化的一个或多个工程化裂解半结构域(也称为二聚结构域突变体),如描述于例如美国专利号8,772,453;8,623,618;8,409,861;8,034,598;7,914,796;和7,888,121中,所有这些专利的公开内容以引用方式全文并入本文。foki位置446、447、479、483、484、486、487、490、491、496、498、499、500、531、534、537以及538处的氨基酸残基全部是影响foki裂解半结构域的二聚的靶标。
形成专性异二聚体的foki的示例性工程化裂解半结构域包括其中第一裂解半结构域包括在foki的位置490和538处的氨基酸残基处的突变,并且第二裂解半结构域包括在氨基酸残基486和499处的突变的对。
因此,在一个实施方案中,490处的突变用lys(k)替换了glu(e);538处的突变用lys(k)替换了iso(i);486处的突变用glu(e)替换了gln(q);并且位置499处的突变用lys(k)替换了iso(i)。具体地,本文所述的工程化裂解半结构域通过在一个裂解半结构域上突变位置490(e→k)和538(i→k)以产生称为“e490k:i538k”的工程化裂解半结构域,并且通过在另一裂解半结构域上突变位置486(q→e)和499(i→l)以产生称为“q486e:i499l”的工程化裂解半结构域来制备。本文所述的工程化裂解半结构域是专性异二聚体突变体,其中异常裂解被被最小化或消除。美国专利号7,914,796和8,034,598,其公开内容以引用方式全文并入。在某些实施方案中,工程化裂解半结构域包含在位置486、499和496(相对于野生型foki编号)处的突变,例如在位置486处用glu(e)残基替换野生型gln(q)残基、在位置499处用leu(l)残基替换野生型iso(i)残基以及在位置496处用asp(d)或glu(e)残基(还分别称为“eld”和“ele”结构域)替换野生型asn(n)残基的突变。在其它实施方案中,工程化裂解半结构域包含在位置490、538和537(相对于野生型foki编号)处的突变,例如在位置490处用lys(k)残基替换野生型glu(e)残基、在位置538处用lys(k)残基替换野生型iso(i)残基以及在位置537处用lys(k)残基或arg(r)残基(还分别称为“kkk”和“kkr”结构域)替换野生型his(h)残基的突变。在其它实施方案中,工程化裂解半结构域包含在位置490和537(相对于野生型foki编号)处的突变,例如在位置490处用lys(k)残基替换野生型glu(e)残基以及在位置537处用lys(k)残基或arg(r)残基(还分别称为“kik”和“kir”结构域)替换野生型his(h)残基的突变。参见例如,美国专利号8,772,453。在其它实施方案中,工程化裂解半结构域包含“sharkey”和/或“sharkey”突变(参见guo等,(2010)j.mol.biol.400(1):96-107)。
本文所述的工程化裂解半结构域可使用任何合适的方法来制备,例如通过如描述于美国专利号7,888,121;7,914,796;8,034,598;和8,623,618中的野生型裂解半结构域(foki)的定点诱变来制备。
可替代地,可使用所谓的“分裂酶”技术在核酸靶位点处体内组装核酸酶(参见例如美国专利公布号20090068164)。此类分裂酶的组分可在单独的表达构建体上表达,或者可在一个开放阅读框中连接,其中各个组分例如被自裂解2a肽或ires序列隔开。组分可以是单个锌指结合结构域或大范围核酸酶核酸结合结构域的结构域。
在使用之前,可例如在如描述于美国专利号8,563,314中的基于酵母的染色体系统中筛选核酸酶的活性。核酸酶的表达可处于组成型启动子或诱导型启动子的控制下,例如在棉子糖和/或半乳糖存在下被激活(去抑制)并在葡萄糖存在下被抑制的半乳糖激酶启动子。
cas9相关的crispr/cas系统包含两个rna非编码组分:反式激活的crrna和含有核酸酶指导序列(间隔区)的前体crrna阵列,所述核酸酶指导序列由相同的直接重复(dr)间隔开。要使用crispr/cas系统完成基因组工程化,必须存在这些rna的两种功能(参见cong等,(2013)sciencexpress1/10.1126/science1231143)。在一些实施方案中,反式激活的crrna和前体crrna通过单独的表达构建体或作为单独的rna来提供。在其它实施方案中,构建嵌合rna,其中将工程化的成熟crrna(赋予靶特异性)融合至反式激活的crrna(提供与cas9的相互作用)以产生嵌合cr-rna-反式激活的crrna杂合体(也称为单指导rna)。(参见jinek,同上和cong,同上)。
如本文所述的核酸酶可在靶位点中进行一个或多个双链和/或单链切割。在某些实施方案中,核酸酶包含催化失活的裂解结构域(例如,foki和/或cas蛋白)。参见美国专利号9,200,266;8,703,489和guillinger等(2014)naturebiotech.32(6):577-582。催化失活的裂解结构域可与催化活性结构域组合作为切口酶,以进行单链切割。因此,可组合使用两种切口酶,以在特定区域中进行双链切割。另外的切口酶也是本领域已知的,例如mccaffery等(2016)nucleicacidsres.44(2):e11.doi:10.1093/nar/gkv878.epub2015年10月19日。
靶位点
如以上详细描述的,可工程化dna结构域以结合至基因座(例如白蛋白或其它安全港基因)中的任何选择序列。与天然存在的dna结合结构域相比,工程化的dna结合结构域可具有新颖的结合特异性。工程化方法包括但不限于合理设计和各种类型的选择。合理设计包括,例如,使用包含三联体(或四联体)核苷酸序列和单个(例如,锌指)氨基酸序列的数据库,其中每个三联体或四联体核苷酸序列与dna结合结构域的一个或多个氨基酸序列相关,所述dna结合结构域结合特定的三联体或四联体序列。参见例如,共有的美国专利6,453,242和6,534,261,其以引用方式全文并入本文。还可进行tal-效应子结构域的合理设计。参见例如,美国公布号20110301073。
适用于dna结合结构域的的示例性选择方法(包括噬菌体展示和双杂交系统)公开于美国专利5,789,538;5,925,523;6,007,988;6,013,453;6,410,248;6,140,466;6,200,759;and6,242,568;aswellaswo98/37186;wo98/53057;wo00/27878;wo01/88197和gb2,338,237中。
靶位点的选择;用于设计和构造融合蛋白(以及编码其的多核苷酸)的核酸酶和方法是本领域技术人员已知的,并且详细描述于美国申请公布号20050064474和20060188987中,其以引用方式全文并入本文。
另外,如在这些和其它参考文献中所公开的,dna结合结构域(例如,多指锌指蛋白)可使用任何合适的接头序列连接在一起,包括例如具有五个或更多个氨基酸的接头。关于示例性6个或更多个氨基酸长度的接头序列,参见例如,美国专利号6,479,626;6,903,185;和7,153,949。本文所述的蛋白质可包括蛋白质的单个dna结合结构域之间的合适接头的任何组合。还参见美国专利号8,586,526。
供体
如上所述,提供了插入外源序列(也称为“供体序列”或“供体”),例如用于校正突变基因或用于增加编码mpsi和/或mpsii疾病中缺少或缺乏的蛋白质(例如,idua或ids)的基因的表达。将显而易见的是,供体序列通常与其被放置处的基因组序列不同。供体序列可含有侧接两个同源区的非同源序列,以允许在感兴趣的位置处进行有效的hdr。另外,供体序列可包含含有与细胞染色质中的感兴趣区域不同源的序列的载体分子。供体分子可含有几个与细胞染色质同源的不连续区域。例如,对于通常不存在于感兴趣区域中的序列的靶向插入,所述序列可存在于供体核酸分子中,并且侧接与感兴趣区域中的序列同源的区域。
本文描述了靶向插入编码idua或ids蛋白的转基因以插入选定位置的方法。用于插入的多核苷酸还可称为“外源”多核苷酸、“供体”多核苷酸或分子或“转基因”。供体多核苷酸可以是单链和/或双链的dna或rna,并且可以线性或环状形式引入细胞中。参见例如,美国专利号8,703,489和9,005,973。供体序列还可包含在dnamc中,其可以环状或线性形式引入细胞中。参见例如,美国专利公布号20140335063。如果以线性形式引入,则供体序列的末端可通过本领域技术人员已知的方法来保护(例如,免受核酸外切降解)。例如,将一个或多个双脱氧核苷酸残基添加至线性分子的3'末端和/或将自互补寡核苷酸连接至一端或两端。参见例如,chang等(1987)proc.natl.acad.sci.usa84:4959-4963;nehls等(1996)science272:886-889。用于保护外源多核苷酸免受降解的另外方法包括但不限于添加末端氨基和使用修饰的核苷酸间键,例如像硫代磷酸酯、氨基磷酸酯和o-甲基核糖或脱氧核糖残基。
可将多核苷酸引入细胞中作为载体分子的一部分,所述载体分子具有另外序列,例如像复制起点、启动子和编码抗生素抗性的基因。此外,供体多核苷酸可作为裸露核酸、作为与试剂诸如脂质体或泊洛沙姆复合的核酸引入,或者可通过病毒(例如,腺病毒、aav、疱疹病毒、逆转录病毒、慢病毒和整合酶缺陷型慢病毒(idlv))递送。
通常插入供体以使得其表达由整合位点处的内源启动子(即驱动其中插入供体的内源基因(例如,高度表达的白蛋白、aavs1、hprt等)的表达的启动子)驱动。然而,将显而易见的是,供体可包含启动子和/或增强子,例如组成型启动子或诱导型或组织特异性启动子。在一些实施方案中,供体在细胞中维持在表达质粒中,使得基因在染色体外表达。
可将供体分子插入内源基因中,使得表达内源基因中的全部、一些或不表达内源基因。例如,可将如本文所述的转基因插入白蛋白或其它基因座中,使得内源白蛋白序列中的一些(编码溶酶体酶的转基因的n-末端和/或c-末端)表达为例如与编码idua或ids蛋白的转基因的融合体或不表达内源白蛋白序列。在其它实施方案中,转基因(例如,有或没有诸如针对白蛋白的另外编码序列)整合到任何内源基因座(例如安全港基因座)中。
当用转基因表达内源序列(内源或转基因的部分)时,内源序列(例如,白蛋白等)可以是全长序列(野生型或突变体)或部分序列。优选地,内源序列是功能性的。这些全长或部分序列(例如,白蛋白)的功能的非限制性实例包括增加由转基因(例如,治疗性基因)表达的多肽的血清半衰期和/或充当载体。
此外,尽管不需要表达,但外源序列还可包括转录或翻译调节序列,例如,启动子、增强子、绝缘子、内部核糖体进入位点、编码2a肽的序列和/或多聚腺苷酸化信号。
在某些实施方案中,外源序列(供体)包含感兴趣蛋白质的融合体和作为其融合配偶体的膜蛋白的细胞外结构域,从而使得融合蛋白位于细胞表面。这允许由转基因编码的蛋白质潜在地在血清中起作用。在治疗mpsi或mpsii疾病的情况下,分别由转基因融合体编码的idua或ids酶从其在细胞(例如,rbc)表面上的位置作用于积聚在血清中的代谢产物。另外,如果rbc被脾脏巨噬细胞按照正常的降解途径吞噬,则在巨噬细胞吞噬所述细胞时形成的溶酶体将在更自然地有利于所述酶的ph值下,使膜结合的融合蛋白暴露于溶酶体中高浓度的代谢产物。
在一些情况下,供体可以是已修饰的内源基因(idua或ids)。例如,可对内源基因进行密码子优化以产生供体。此外,尽管抗体对酶替代疗法的响应相对于所讨论的特定治疗性酶和个体患者而变化,但在用野生型idua或ids进行酶替代治疗的许多mpsi或mpsii疾病患者中已观察到显著的免疫应答。另外,这些抗体与治疗功效的相关性也是可变的(参见katherineponder,(2008)jclininvest118(8):2686)。因此,与野生型idua或ids相比,本发明的方法和组合物可包括具有修饰序列的供体的产生,包括但不限于在已知为内源免疫应答引发表位的位点处产生功能性沉默氨基酸变化的修饰,和/或使得由这种供体产生的多肽免疫原性较低的截短。
由于缺少脑中缺失的idua或ids酶,mpsi和mpsii疾病患者经常具有神经后遗症。不幸的是,由于血脑屏障的不可渗透性,经常难以通过血液向脑递送治疗剂。因此,本发明的方法和组合物可与增加治疗剂向脑中递送的方法结合使用,包括但不限于导致脑毛细血管的细胞之间紧密连接瞬间打开的方法,诸如通过使用颈动脉内施用高渗甘露醇溶液进行瞬间渗透性破坏、使用聚焦超声以及施用缓激肽类似物。可替代地,可设计治疗剂以利用受体或转运机制用于特异性转运至脑中。可使用的特异性受体的实例包括转铁蛋白受体、胰岛素受体或低密度脂蛋白受体相关蛋白1和2(lrp-1和lrp-2)。已知lrp与一系列分泌性蛋白质诸如apoe、tpa、pai-1等相互作用,并且因此将来自这些蛋白质之一的识别序列与lrp融合可能有利于在肝脏中表达治疗性蛋白质和分泌到血流中后将酶转运到脑中(参见gabathuler,(2010)同上)。
细胞
本文还提供了基因修饰的细胞,例如包含编码idua和/或ids蛋白的转基因的肝细胞或干细胞,包括通过本文所述方法产生的细胞。idua和/或ids转基因可在染色体外表达,或者可使用一种或多种核酸酶以靶向方式整合到细胞的基因组中。与随机整合不同,核酸酶介导的靶向整合确保转基因整合到特定基因中。转基因可整合到靶基因中的任何位置。在某些实施方案中,转基因在核酸酶结合和/或裂解位点处或附近整合,例如,在裂解和/或结合位点上游或下游的1-300(或其间的任何数量的碱基对)基对内,更优选地在裂解和/或结合位点任一侧的1-100(或其间的任何数量的碱基对)个碱基对内,甚至更优选地在裂解和/或结合位点任一侧的1至50(或其间的任何数量的碱基对)个碱基对内。在某些实施方案中,整合的序列不包括任何载体序列(例如,病毒载体序列)。
可如本文所述对任何细胞类型(包括但不限于细胞或细胞系)进行基因修饰以包含转基因。如本文所述的基因修饰细胞的其它非限制性实例包括t细胞(例如,cd4+、cd3+、cd8+等);树突状细胞;b细胞;自体的(例如,源自患者的)。在某些实施方案中,所述细胞是肝细胞并且在体内被修饰。在某些实施方案中,所述细胞是干细胞,包括异源多能干细胞、全能干细胞或多能干细胞(例如,cd34+细胞、诱导多能干细胞(ipsc)、胚胎干细胞等)。在某些实施方案中,如本文所述的细胞是源自患者的干细胞。
如本文所述的细胞可用于例如通过体内疗法治疗和/或预防患有所述病症的受试者中的mpsi或mpsii疾病。还提供了离体疗法,例如在核酸酶修饰的细胞可扩增并且然后使用标准技术重新引入患者体内的情况下。参见例如,tebas等(2014)newengjmed370(10):901。在干细胞的情况下,在输注到受试者体内后,也发生这些前体体内分化成表达功能性蛋白质(来自插入的供体)的细胞。
还提供了包含如本文所述的细胞的药物组合物。另外,所述细胞可在向患者施用之前冷冻保存。
递送
核酸酶、编码这些核酸酶的多核苷酸、供体多核苷酸和包含本文所述蛋白质和/或多核苷酸的组合物可通过任何合适的方式体内或离体递送。
递送如本文所述的核酸酶的方法描述于例如美国专利号6,453,242;6,503,717;6,534,261;6,599,692;6,607,882;6,689,558;6,824,978;6,933,113;6,979,539;7,013,219;和7,163,824中,所有这些专利的公开内容以引用方式全文并入本文。
还可使用含有编码锌指、talen和/或cas蛋白中的一种或多种的序列的载体递送如本文所述的核酸酶和/或供体构建体。可使用任何载体系统,包括但不限于质粒载体、逆转录病毒载体、慢病毒载体、腺病毒载体、痘病毒载体;疱疹病毒载体以及腺相关病毒载体等。还参见美国专利号6,534,261;6,607,882;6,824,978;6,933,113;6,979,539;7,013,219;和7,163,824,其以引用方式全文并入本文。此外,将显而易见的是,这些载体中的任一种可包含治疗所需的一种或多种序列。因此,当将一种或多种核酸酶和供体构建体引入细胞时,核酸酶和/或供体多核苷酸可携带在相同载体上或不同载体上。当使用多个载体时,每个载体可包含编码一种或多种核酸酶和/或供体构建体的序列。
常规基于病毒和非病毒的基因转移方法可用于将编码核酸酶和供体构建体的核酸引入细胞(例如,哺乳动物细胞)和靶组织中。非病毒载体递送系统包括dna质粒、裸露核酸和与递送载体诸如脂质体或泊洛沙姆复合的核酸。病毒载体递送系统包括dna和rna病毒,其在递送至细胞后具有游离或整合的基因组。关于基因疗法程序的综述,参见anderson,science256:808-813(1992);nabel和felgner,tibtech11:211-217(1993);mitani&caskey,tibtech11:162-166(1993);dillon,tibtech11:167-175(1993);miller,nature357:455-460(1992);vanbrunt,biotechnology6(10):1149-1154(1988);vigne,restorativeneurologyandneuroscience8:35-36(1995);kremer和perricaudet,britishmedicalbulletin51(1):31-44(1995);haddada等,incurrenttopicsinmicrobiologyandimmunologydoerflerand
非病毒递送核酸的方法包括电穿孔、脂转染、显微注射、基因枪、病毒体、脂质体、免疫脂质体、聚阳离子或脂质:核酸缀合物、裸露dna、人工病毒粒子和dna的试剂增强摄取。使用例如,sonitron2000系统(rich-mar)的声孔效应也可用于递送核酸。
另外的示例性核酸递送系统包括由amaxabiosystems(cologne,germany)、maxcyte公司(rockville,maryland)、btxmoleculardeliverysystems(holliston,ma)和copernicustherapeutics公司提供的那些(参见例如us6008336)。脂转染描述于例如,美国专利号5,049,386;4,946,787;和4,897,355中,并且脂转染试剂是商业销售的(例如,transfectamtm和lipofectintm)。适用于多核苷酸的有效受体识别脂转染的阳离子和中性脂质包括felgner、wo91/17424、wo91/16024的那些。
脂质:核酸复合物(包括靶向脂质体,诸如免疫脂质复合物)的制备是本领域技术人员熟知的(参见例如,crystal,science270:404-410(1995);blaese等,cancergenether.2:291-297(1995);behr等,bioconjugatechem.5:382-389(1994);remy等,bioconjugatechem.5:647-654(1994);gao等,genetherapy2:710-722(1995);ahmad等,cancerres.52:4817-4820(1992);美国专利号4,186,183、4,217,344、4,235,871、4,261,975、4,485,054、4,501,728、4,774,085、4,837,028和4,946,787)。
另外的递送方法包括使用将待递送的核酸包装至engeneic递送载体(edv)中。使用双特异性抗体将这些edv特异性递送至靶组织,其中所述抗体的一个臂对靶组织具有特异性,并且另一个臂对edv具有特异性。抗体将edv带到靶细胞表面,并且然后通过胞吞作用将edv带入细胞中。一旦进入细胞中,则释放内容物(参见macdiarmid等(2009)naturebiotechnology27(7):643)。
使用基于rna或dna病毒的系统递送编码工程化zfp的核酸利用用于使病毒靶向体内特定细胞并且使病毒有效载荷运输至核中的高度进展的方法。病毒载体可直接向受试者施用(体内)或它们可用于体外处理细胞并且向受试者施用修饰的细胞(离体)。用于递送zfp的常规基于病毒的系统包括但不限于用于基因转移的逆转录病毒、慢病毒、腺病毒、腺相关、牛痘和单纯疱疹病毒载体。用逆转录病毒、慢病毒和腺相关病毒基因转移方法有可能在宿主基因组中达成整合,从而经常导致插入的转基因的长期表达。另外,已在许多不同细胞类型和靶标组织中观察到高转导效率。
逆转录病毒的向性可通过掺入外来包膜蛋白来改变,从而扩展靶细胞的潜在靶标群。慢病毒载体是能够转导或感染非分裂细胞并且通常产生高病毒效价的逆转录病毒载体。逆转录病毒基因转移系统的选择取决于靶组织。逆转录病毒载体由具有高达6-10kb外来序列的包装能力的顺式作用长末端重复组成。最小顺式作用ltr足以用于载体的复制和包装,然后将其用于将治疗性基因整合到靶细胞中以提供永久性转基因表达。广泛使用的逆转录病毒载体包括基于鼠白血病病毒(mulv)、长臂猿白血病病毒(galv)、猿猴免疫缺陷病毒(siv)、人免疫缺陷病毒(hiv)及其组合的那些载体(参见例如,buchscher等,j.virol.66:2731-2739(1992);johann等,j.virol.66:1635-1640(1992);sommerfelt等,virol.176:58-59(1990);wilson等,j.virol.63:2374-2378(1989);miller等,j.virol.65:2220-2224(1991);pct/us94/05700)。
在其中优选瞬间表达的应用中,可使用基于腺病毒的系统。基于腺病毒的载体能够在许多细胞类型中具有非常高的转导效率并且不需要细胞分裂。用此类载体已获得了高效价和高表达水平。此载体可在相对简单系统中大量产生。腺相关病毒(“aav”)载体还用于例如在核酸和肽的体外产生中用靶核酸转导细胞,以及用于体内和离体基因疗法程序(参见例如,west等,virology160:38-47(1987);美国专利号4,797,368;wo93/24641;kotin,humangenetherapy5:793-801(1994);muzyczka,j.clin.invest.94:1351(1994)。重组aav载体的构建描述于许多出版物中,包括美国专利号5,173,414;tratschin等,mol.cell.biol.5:3251-3260(1985);tratschin等,mol.cell.biol.4:2072-2081(1984);hermonat和muzyczka,pnas81:6466-6470(1984);和samulski等,j.virol.63:03822-3828(1989)。
目前临床试验中至少有六种病毒载体方法可用于基因转移,其利用涉及通过插入辅助细胞系中的基因互补缺陷载体以产生转导剂的方法。
plasn和mfg-s是已用于临床试验的逆转录病毒载体的实例(dunbar等,blood85:3048-305(1995);kohn等,nat.med.1:1017-102(1995);malech等,pnas94:2212133-12138(1997))。pa317/plasn是基因疗法试验中使用的第一种治疗性载体。(blaese等,science270:475-480(1995))。已观察到mfg-s包装载体的转导效率为50%或更高。(ellem等,immunolimmunother.44(1):10-20(1997);dranoff等,hum.genether.1:111-2(1997)。
重组腺相关病毒载体(raav)是基于缺陷性和非病原性细小病毒腺相关2型病毒的有前途的替代性基因递送系统。所有载体都源自仅保留侧接转基因表达盒的aav145bp倒置末端重复序列的质粒。归因于整合到转导的细胞的基因组中的高效基因转移和稳定转基因递送是此载体系统的关键特征。(wagner等,lancet351:91171702-3(1998),kearns等,genether.9:748-55(1996))。还可根据本发明使用其它aav血清型,包括非限制性实例,aav1、aav3、aav4、aav5、aav6、aav8、aav8.2、aav9和aavrh10以及假型aav诸如aav2/8、aav2/5和aav2/6。
复制缺陷型重组腺病毒载体(ad)可在高效价下产生并且易于感染许多不同的细胞类型。大多数腺病毒载体被工程化使得转基因替换ade1a、e1b和/或e3基因;随后使复制缺陷性载体在反式提供缺失的基因功能的人293细胞中增殖。ad载体可在体内转导多种类型的组织,包括非分裂性分化细胞,诸如在肝脏、肾脏和肌肉中发现的那些细胞。常规ad载体具有大携带能力。在临床试验中使用ad载体的实例涉及用于肌内注射的抗肿瘤免疫的多核苷酸疗法(sterman等,hum.genether.7:1083-9(1998))。在临床试验中使用腺病毒载体进行基因转移的其它实例包括rosenecker等,infection24:15-10(1996);sterman等,hum.genether.9:71083-1089(1998);welsh等,hum.genether.2:205-18(1995);alvarez等,hum.genether.5:597-613(1997);topf等,genether.5:507-513(1998);sterman等,hum.genether.7:1083-1089(1998)。
包装细胞用于形成能够感染宿主细胞的病毒颗粒。此类细胞包括包装腺病毒的293细胞和包装逆转录病毒的ψ2细胞或pa317细胞。用于基因疗法的病毒载体通常由将核酸载体包装入病毒颗粒中的生产细胞系产生。载体通常含有包装和随后整合到宿主中(如果适用)所需的最小病毒序列,其它病毒序列被编码待表达蛋白质的表达盒替换。缺失的病毒功能由包装细胞系反式提供。例如,用于基因疗法的aav载体通常仅具有来自aav基因组的倒置末端重复(itr)序列,其是包装和整合到宿主基因组中所需的。病毒dna被包装在细胞系中,所述细胞系含有编码其它aav基因的辅助质粒,即rep和cap,但缺少itr序列。所述细胞系还被作为辅助病毒的腺病毒感染。辅助病毒促进aav载体的复制和来自辅助质粒的aav基因的表达。由于缺少itr序列,辅助质粒未以显著量包装。腺病毒的污染可通过例如,腺病毒比aav更敏感的热处理来减少。
在许多基因疗法应用中,希望的是基因疗法载体以高度特异性递送至特定组织类型。因此,通过在病毒外表面上用病毒外壳蛋白表达配体作为融合蛋白,病毒载体可被修饰以对给定细胞类型具有特异性。选择配体以对已知存在于感兴趣细胞类型上的受体具有亲和力。例如,han等,proc.natl.acad.sci.usa92:9747-9751(1995)报道了moloney鼠白血病病毒可被修饰以表达与gp70融合的人调蛋白,并且重组病毒感染某些表达人表皮生长因子受体的人乳腺癌细胞。此原理可扩展到其它病毒-靶细胞对,其中靶细胞表达受体并且病毒表达包含细胞表面受体配体的融合蛋白。例如,可工程化丝状噬菌体以展示对几乎任何选择的细胞受体具有特异性结合亲和力的抗体片段(例如,fab或fv)。尽管以上描述主要适用于病毒载体,但相同的原理可应用于非病毒载体。可将此类载体工程化以含有特异性摄取序列,其有利于特定靶细胞的摄取。
如下所述,基因疗法载体可通常通过全身性施用(例如,静脉内、腹膜内、肌内、真皮下或颅内输注)或局部施用,通过向个体患者施用来体内递送。可替代地,载体可离体递送至细胞,诸如从个体患者外植的细胞(例如,淋巴细胞、骨髓吸出物、组织生检体)或通用供体造血干细胞中,随后通常在选择已并入载体的细胞之后将细胞再植入患者中。
含有核酸酶和/或供体构建体的载体(例如,逆转录病毒、腺病毒、脂质体等)还可直接向生物体施用以在体内转导细胞。可替代地,可施用裸露dna。通过通常用于使分子与血液或组织细胞最终接触的任何途径进行施用,包括但不限于注射、输注、局部施用和电穿孔。施用此类核酸的合适方法是可用的且为本领域技术人员所熟知,并且尽管多于一种途径可用于施用特定组合物,但特定途径可经常提供比另一途径更即刻且更有效的反应。
适用于引入本文所述多核苷酸的载体包括非整合型慢病毒载体(idlv)。参见例如,ory等(1996)proc.natl.acad.sci.usa93:11382-11388;dull等(1998)j.virol.72:8463-8471;zuffery等(1998)j.virol.72:9873-9880;follenzi等(2000)naturegenetics25:217-222;美国专利公布号2009/054985。
药学上可接受的载体部分地通过所施用的具体组合物以及通过用于施用组合物的具体方法来确定。因此,如下所述,药物组合物存在广泛多种的合适制剂(参见例如,remington’spharmaceuticalsciences,第17版,1989)。
将显而易见的是,核酸酶编码序列和供体构建体可使用相同或不同的系统递送。例如,供体多核苷酸可由质粒携带,而一种或多种核酸酶可由aav载体携带。此外,不同的载体可通过相同或不同的途径施用(肌内注射、尾静脉注射、其它静脉内注射、腹膜内施用和/或肌内注射)。载体可同时或以任何顺序递送。
用于离体和体内施用两者的制剂包括液体或乳化液体中的混悬剂。活性成分经常与药学上可接受并与活性成分相容的赋形剂混合。合适的赋形剂包括例如水、盐水、右旋糖、甘油、乙醇等或其组合。另外,组合物可含有少量辅助物质,诸如润湿剂或乳化剂、ph缓冲剂、稳定剂或增强药物组合物有效性的其它试剂。
应用
本发明的方法考虑治疗和/或预防mpsi疾病(例如溶酶体贮积病)和/或mpsi疾病。治疗可包括将一种或多种校正性疾病相关基因(例如,idua、ids等)插入细胞中的安全港基因座(例如白蛋白)内以表达所需的酶并释放到血流中。转基因可编码蛋白质,其包含密码子优化的转基因(例如,idua或ids);和/或其中可去除表位而不在功能上改变蛋白质的转基因。在一些情况下,所述方法包括将表达校正性酶编码转基因的游离基因插入细胞中以表达所需的酶并释放到血流中。插入分泌细胞(诸如肝细胞)中以将产物释放到血流中是特别有用的。本发明的方法和组合物还可用于其中希望在造血干细胞中提供编码一种或多种治疗剂的idua或ids转基因使得衍生自这些细胞的成熟细胞(例如,rbc)含有治疗剂的任何情况。这些干细胞可在体外或体内分化,并且可衍生自可用于所有患者的通用供体类型的细胞。另外,所述细胞可含有跨膜蛋白以在体内运输细胞。治疗还可包括使用含有治疗性转基因的患者细胞,其中细胞离体发育,并且然后将其引回患者体内。例如,可通过骨髓移植将含有合适的idua或ids编码转基因的hsc插入患者体内。可替代地,已使用idua或ids编码转基因编辑的干细胞诸如肌肉干细胞或ipsc也可注射到肌肉组织中。
因此,此技术可用于其中患者由于问题(例如,表达水平的问题或蛋白质表达为亚功能或无功能的问题)而缺乏某些蛋白质的情况。对本发明特别有用的是表达转基因以校正或恢复患有mpsi疾病的受试者的功能。
作为非限制性实例,完成缺陷或缺失蛋白质的产生并用于治疗mpsi和/或mpsii疾病。编码蛋白质的核酸供体可插入安全港基因座(例如白蛋白或hprt)中,并使用外源启动子或使用存在于安全港处的启动子表达。可替代地,供体可用于原位校正缺陷基因。可将希望的idua或ids编码转基因插入cd34+干细胞中并在骨髓移植期间返回至患者。最后,可将核酸供体插入β珠蛋白基因座处的cd34+干细胞中,使得衍生自所述细胞的成熟红细胞具有由核酸供体编码的高浓度生物制剂。然后可通过跨膜蛋白(例如受体或抗体)将含有生物制剂的rbc靶向正确的组织。另外,rbc可通过电致敏化离体致敏,以使它们在暴露于能量源后更容易破裂(参见wo2002007752)。
在一些应用中,可通过使用本发明的方法和组合物敲除内源基因。这个方面的实例包括敲除异常基因调节子或异常疾病相关基因。在一些应用中,异常内源基因可在功能上或原位上用所述基因的野生型型式替换。还可改变插入的基因以改善治疗性idua或ids蛋白的表达或降低其免疫原性。在一些应用中,插入的idua或ids编码转基因是融合蛋白,以增加其向选定组织(诸如脑)的转运。
以下实施例涉及本公开的示例性实施方案,其中核酸酶包含锌指核酸酶(zfn)或talen。应理解,这仅出于举例说明的目的,并且可使用其它核酸酶或核酸酶系统,例如具有工程化dna结合结构域的归巢内切核酸酶(大范围核酸酶)和/或天然存在的工程化归巢内切核酸酶(大范围核酸酶)dna结合结构域和异源裂解结构域的融合体和/或包含工程化单指导rna的crispr/cas系统。
实施例
实施例1:设计和构建小鼠zfn试剂和idua供体模板
由于临床候选zfn和人白蛋白供体同源臂的靶序列在小鼠白蛋白基因座中不是保守的,因此开发了小鼠替代试剂用于这些小鼠研究。小鼠替代试剂包括靶向小鼠白蛋白基因的内含子1中的同源位点的一对zfnraav载体(sb-48641、sb-31523,如下表i和表ii所示,参见美国专利申请14/872,537),以及编码侧接与小鼠基因座具有同源性的臂的无启动子人idua转基因(hidua)的供体(sb-mu-idua)。本研究中使用的zfn:zfn:供体的比率为1:1:8。对于此小鼠研究,使用血清型raav2/8包装和递送替代试剂,因为其在小鼠体内赋予比raav2/6载体更好的转导和更快的转基因表达。将raav2/8载体稀释到制剂缓冲液,补充有35mmnacl和5%甘油,ph7.1的磷酸盐缓冲盐水(pbs)中。
表1:小鼠白蛋白设计
表2:小鼠白蛋白特异性锌指的靶位点
小鼠白蛋白特异性zfn对具有完全活性。
实施例2:在小鼠模型中治疗mpsi(idua敲除或idua-/-)
a.材料和方法
本研究中使用的mpsi(idua敲除或idua-/-)小鼠模型由elizabethneufeld博士(ucla,losangeles,ca)通过破坏idua基因产生,其中新霉素抗性盒被插入外显子6中(ohmi等(2003).proc.natl.acad.sci.usa100(4):1902–1907)。然后将小鼠繁殖到c57bl/6遗传背景上,并选择杂合子以产生idua-/-小鼠。这种纯合突变在动物的生命过程中导致显著的形态学、生物化学和神经学变化,并且已被证明是用于溶酶体病理生理学研究的合适模型。特别地,这些mpsi小鼠已证明表现出典型的mpsi表型特征,诸如颅面畸形、神经功能缺陷以及组织和尿液中gag水平升高(hartung等(2004).molther.9(6):866-75;ou等(2014).molgenetmetab.111(2):116-22)。idua-/-小鼠在约3-5周龄后开始出现这些症状,并且到12-16周龄时gag的大量溶酶体积聚在肝脏和中枢神经系统(cns,即脑、脊髓和脑膜)中很明显。另外,这些mpsi小鼠显示出认知缺陷/损伤(hartung等,同上;ou等,同上)。
当幼仔变得可用时,将小鼠组随机分组并且在4-10周龄之间给药。每组至少2只小鼠在同一天给药。
给动物施用环磷酰胺以抑制对hidua蛋白的可能的免疫原性应答。所有小鼠在给药前一天接受200μl腹膜内(ip)注射环磷酰胺(120mg/kg),并且然后每周一次直至第12周。在第12周后,小鼠每两周一次接受120mg/kg直至尸检。在第12周后环磷酰胺施用频率变化的原因是由于在少数小鼠中观察到在注射部位附近出现轻度脱毛和急性轻度运动迟缓,这是环磷酰胺的已知副作用。先前的研究已证明使用双月注射方案(aronovich等(2007).j.genemed.9:403-15)或根据需要(参见ou等(2014)molgenetmetabol111(2):116)有效免疫抑制以减轻mpsi小鼠中人idua活性的丧失。
基于每个性别的年龄和体重随机分配动物。在第1天施用单剂量的测试物或载体。在aav注射后1个月,将每组的三只小鼠安乐死。在aav注射后4个月,将每组剩余的5只小鼠安乐死。zfn+供体组的总raav2/8剂量水平为l.5el2vg/只小鼠,而仅供体组的总raav2/8剂量水平为l.2e12vg/只小鼠。接受的组和剂量如下表3所示。
表3:组名称和剂量水平
收集和测量:在尸检(第120天)之前通过颌下出血从所有动物收集用于血液学和临床化学分析的血液样品。在血清化学收集之前,使动物禁食至少8小时。将用于血液学分析的血液样品(目标体积100-150μl)收集在含有钾(k3)edta作为抗凝血剂的管中。将用于临床化学分析的血液样品(目标体积100-150μl)收集在没有抗凝血剂的管中并加工成血清。
为了评价idua活性,在第7、14、28、60、90、104和120天通过来自所有小鼠的颌下出血将血液样品(目标体积200μl)收集在含有肝素作为抗凝血剂的管中并加工成血浆。
为了评价尿糖胺聚糖(gag)水平,在第7、14、21、28、42、60、74、90、104和120天,通过向膀胱轻轻施加压力,从所有小鼠收集尿样品(大约10-100μl)。
从小鼠组织的基因组dna分离:将大约5mg冷冻组织置于裂解基质(lysismatrix)d管(mpbiomedicals,burlingame,ca)中,并加入400μl组织和细胞裂解液(epicentre,madison,wi)。均化在
基因修饰分析:miseq深度测序:为了评估靶向肝细胞中小鼠替代zfn构建体的活性,从每只小鼠的肝脏中分离基因组dna。使用miseq系统(illumina,sandiegoca)使zfn靶位点经受序列分析。设计一对寡核苷酸引物用于扩增跨越小鼠白蛋白基因座中的zfn靶位点的169碱基对(bp)片段(表4),并引入用于第二轮扩增的结合序列(粗体)。纯化此聚合酶链反应(pcr)扩增的产物,并用设计用于引入扩增子特异性标识符序列(“条形码”)的寡核苷酸以及设计用于结合测序寡核苷酸引物的末端区域使其经受第二轮pcr。然后使条形码扩增子的混合群经受miseq分析,这是一种固相测序程序,其允许在单个测定芯片上平行分析数千个样品。miseq方案对每个输入寡核苷酸顺序进行正向和反向两种测序反应。将配对序列比对并合并,并且与野生型序列比较以对插入和/或缺失(“插入缺失”)作图。zfn活性报告为“插入缺失%”,即由于插入或缺失而与野生型不同的测序扩增子的分数。
表4.用于miseq插入/缺失(插入缺失)分析的寡核苷酸
hidua蛋白质印迹:为了检查hidua在小鼠肝细胞中的表达,对肝蛋白提取物进行蛋白质印迹。将大约50mg冷冻组织置于裂解基质d管(mpbiomedicals,burlingame,ca)中,并加入500μl补充有halt蛋白酶抑制剂(thermofisherscientific)的ripa缓冲液。均化在
idua酶活性测定:使用合成底物4-甲基伞形酮基α-l-艾杜糖醛酸苷(4-mu-艾杜糖醛酸苷;glycosynth,warrington,cheshire,england)在荧光测定法中评价血浆和组织蛋白提取物中的idua酶活性。通过将小鼠组织样品与1mlpbs(ph7.4)混合来制备组织蛋白提取物。将样品储存在冰上,并用
在idua酶活性测定中,将25μl小鼠组织蛋白提取物或小鼠血浆样品与25μl溶解在甲酸盐缓冲液(0.4m甲酸钠,ph3.5,0.2%tritonx-100)中的360μm合成底物(4-mu-艾杜糖醛酸苷)混合。将反应物在37℃下孵育30分钟,并通过加入200μl甘氨酸碳酸盐缓冲液(84mm甘氨酸、85mm无水碳酸钠)终止反应。通过使用biotek酶标仪(biotek,winooski,vt)测量荧光(ex365/em450)测定4-mu的释放。使用4-mu稀释液(sigma-aldrich,st.louis,mo)产生标准曲线。将所得数据用线性曲线拟合,并使用此最佳拟合曲线计算测试样品中4-mu的浓度。将酶活性表示为每小时测定孵育时间、每ml血浆或每mg组织提取物释放的nmol4-mu(nmol/小时/ml或nmol/小时/mg)。
评价尿和小鼠组织中的gag水平:根据制造商的说明书,使用blyscantm糖胺聚糖测定试剂盒(biocolor,carrickfergus,countyantrim,unitedkingdom)测定组织和尿中的gag水平。在gag测定之前,将如上所述制备的小鼠组织匀浆与蛋白酶k(20mg/ml)以1:3的比率混合,并在55℃下消化24小时,随后通过煮沸10分钟使蛋白酶k热灭活。用200单位的dna酶和2μg的rna酶在室温下再次消化组织匀浆24小时,同时摇动。然后通过煮沸10分钟使dna酶和rna酶热灭活,并将所得组织匀浆用于gag测定。
组织病理学分析:在第120天的雄性(第1组[n=5],第2组[n=4],第4组[n=4]和第6组[n=2])和雌性(第3组[n=4],第5组[n=5]和第6组[n=2])尸检时收集终末身体和器官(肾上腺、脑、心脏、肾脏、肝脏、脾脏、睾丸或卵巢)的重量,并且通常在excel中计算组平均值和标准偏差。
对第1组至第6组的动物进行尸检,并将收集的组织置于10%中性缓冲福尔马林(nbf)中进行固定。修剪以下组织以产生组织学切片:肾上腺、主动脉、脑、盲肠、子宫颈、结肠、十二指肠、附睾、食道、眼、股骨-胫骨关节、胆囊、哈氏腺、心脏、回肠、注射部位、空肠、肾脏、喉、肝脏、肺与支气管、淋巴结(肠系膜)、视神经、卵巢、胰腺、甲状旁腺、脑垂体、前列腺、直肠、唾液腺、坐骨神经、精囊、骨骼肌(股四头肌)、皮肤、脊髓(颈椎、腰椎、胸椎)、脾脏、胸骨与骨髓、胃、睾丸、胸腺、甲状腺、舌、气管、膀胱、子宫和阴道。对来自所有剂量组的动物的上述组织进行常规处理,并包埋在石蜡块中。将块切片并用苏木精和曙红(h&e)染色。由经过委员会认证的兽医病理学家在显微镜下评估载玻片。
b.结果
基因修饰分析:白蛋白基因座处的插入缺失百分比:为了评估体内靶向肝细胞中zfn构建体的活性,在尸检时从所有小鼠的肝脏中分离基因组dna。使用miseq系统(illumina,sandiegoca)使小鼠白蛋白内含子1基因座处的zfn靶位点经受序列分析。然后通过小插入和/或缺失(插入缺失)的存在(其是zfn活性的标志)来确定zfn活性。在接受替代小鼠zfn+sb-mu-idua的所有小鼠组(第4和第5组)中检测到zfn活性。1个月时肝组织中白蛋白基因修饰的水平(插入缺失%)对于雄性和雌性动物分别为46.67±5.74%和34.05±2.06%(平均值±sd)。4个月时基因修饰的水平对于雄性和雌性动物分别为55.65±4.08%和45.69±2.26%。在任一时间点,在制剂缓冲液对照组或仅供体组中没有zfn切割水平高于背景。
为了证实肝脏特异性启动子的特异性,还对来自脾脏、心脏和肌肉的基因组dna进行了miseq分析。选择这些器官,因为它们在zfn+供体治疗的小鼠中显示出肝脏外最高水平的idua活性(参见图1a)。在第4组和第5组的小鼠上进行这些分析,因为它们在靶组织上显示出最高水平的zfn活性。包括相应的未治疗的雄性小鼠(第2组)作为阴性对照。4个月时在第2组和第4组的脾脏和心脏(非靶组织)中未观察到基因修饰,这证实了肝脏特异性启动子的特异性,并且证明了在这些组织中发现的idua活性和gag降低不是源自肝脏外的zfn活性和供体整合。
hidua蛋白质印迹。在确认肝细胞中小鼠白蛋白基因座的适当基因修饰后,接着检查来自肝脏的蛋白质提取物的白蛋白基因座的hidua表达。为了避免来自内源性小鼠idua的潜在背景信号,使用针对人idua产生的检测抗体进行蛋白质印迹分析。
如图1b所示,在1和4个月时在接受小鼠替代zfn+hidua供体的所有雄性和雌性小鼠的肝脏中可检测到hidua。表达似乎在4个月时增加了。注射单独的制剂缓冲液的野生型对照小鼠未显示hidua信号,这证明了所用抗体的物种特异性。hidua的表达取决于zfn和hidua供体两者的存在,因为在不存在zfn(仅供体组)的情况下未观察到hidua信号。
idua酶活性。为了确定由肝脏白蛋白基因座表达的hidua转基因是否产生了功能活性蛋白,对于idua进行基于荧光的酶活性测定。此测定在1)来自肝脏的蛋白质提取物上进行(以确保表达的酶具有活性);2)血浆上进行(以确定表达的酶是否由肝细胞分泌);3)来自脑、心脏、肺、肌肉和脾脏的蛋白质提取物上进行(以确定酶是否以能够被二级组织摄取的形式表达)。
在1个月时,zfn+供体治疗的小鼠肝脏中的idua活性(对于雄性和雌性动物分别为65.26±24.68和40.54±14.54nmol/小时/mg)比在野生型小鼠中发现的活性水平(4.09±0.99nmol/小时/mg)高9至10倍。与对照(0.11±0.02[雄性]和0.16±0.04[雌性]nmol/小时/mg)和仅供体(0.18±0.05nmol/小时/mg)mpsi小鼠两者中的活性相比,zfn+供体治疗的小鼠中的idua活性也显著增加了。在不存在zfn(仅供体)的情况下接受sb-mu-idua供体的mpsi小鼠中发现的低水平活性证明在小鼠肝细胞中整合和表达hidua需要zfn和sb-mu-idua供体两者。
在4个月时,与野生型动物中的活性(8.05±1.55nmol/小时/mg)相比,zfn+供体治疗的小鼠中肝脏idua活性进一步增加(对于雄性和雌性动物分别为147.32±65.86和63.59±43.42nmol/小时/mg)。
如图2a所示,从第14天开始和整个研究中,与在对照和仅供体小鼠中观察到的水平相比,zfn+供体治疗的小鼠中的血浆idua酶活性也增加了。从第14天直至研究结束,平均idua活性保持高于野生型对照小鼠中观察到的内源性生理水平的约1.5-10倍。在1个月和4个月时在脑、心脏、肝脏、肺、肌肉和脾脏中比较idua活性(图2c),并且在4个月时在心脏、肝脏、肺和脾脏(雄性和雌性)以及肌肉(仅雄性)中,zfn+供体组中的idua活性显示出比性别匹配的未治疗mpsi小鼠显著更高的活性。
在接受替代小鼠zfn+hidua供体两者的mpsi小鼠中,在1个月时除了脑之外的所有非靶组织中的idua活性接近野生型小鼠中的活性水平,这证明了在肝细胞中白蛋白基因座处产生的idua被分泌到血浆中,其以活性形式被二级组织从血浆摄取(参见,图4)。在脑中,检测到idua活性的增加,这对应于在野生型小鼠的脑中发现的大约5%的idua活性(参见,图4e)。
评价尿和小鼠组织中的gag水平:为了确定在zfn+供体治疗的mpsi小鼠中观察到的idua酶缺乏的校正是否与防止疾病生物标志物糖胺聚糖(gag)的水平随时间推移升高或减少相关,在所有动物的尿和组织提取物中测定gag水平。
结果指示,与野生型小鼠中的水平相比,在接受制剂缓冲液的mpsi小鼠中,随着疾病进展,尿gag水平在整个研究中增加了。如图3c所示,在给药后第14天,在雄性和雌性mpsi小鼠中用替代小鼠zfn和hidua供体两者进行治疗防止了在未治疗mpsi小鼠中所见的尿gag水平的增加。与野生型小鼠相比,配制缓冲液治疗的雄性和雌性mpsi小鼠的所有组织(脑除外)中的gag水平也显著增加了(参见图5)。这些水平在zfn+供体治疗的mpsi动物中完全正常化。在肝脏(其中idua由白蛋白基因座产生)和二级组织(其必须从血浆摄取idua)两者中都发生正常化。在脑中,与未治疗的动物相比,治疗的动物中的gag水平较低(参见,图5e)。
此数据显示肝脏产生的hidua进入循环(血浆)并被脾脏、心脏、肝脏、肺和骨骼肌摄取,并导致zfn+供体治疗的mpsi小鼠的二级组织中gag水平降低。
组织病理学评价
临时和终末尸检,研究的第28天和第120天
从第28天尸检评估组织的子组(脑、肺、心脏、肝脏、骨骼肌和脾脏)。
野生型c57bl/6对照动物未显示与测试物相关的病理学发现。雄性和雌性mpsi对照小鼠在整个身体中形成各种细胞类型的空泡化,并且这被解释为糖胺聚糖(gag)的溶酶体积聚。空泡化是充满糖胺聚糖的溶酶体的特征性微观外观(ohmi等(2003)procnatlacadsciusa100(4):1902-1906)。当与第28天的雄性和雌性mpsi对照小鼠比较时,第120天的动物在整个身体中具有增加的各种细胞类型典型空泡化的发生率和/或严重性,其被认为是gag的溶酶体积聚。野生型c57bl/6对照动物在第28天或第120天未显示与测试物相关的病理学影响。
cns组织评估。当与mpsi对照比较时,用zfn+hidua供体或仅hidua供体治疗的动物在脑中具有相似的大神经元或神经胶质空泡化的发生率和/或严重性。然而,在脊髓中,给予zfn+hidua供体的雄性具有降低的神经元或神经胶质空泡化的发生率和/或严重性。同样,除了雌性腰脊髓中的神经胶质空泡化,给予zfn+hidua供体的雌性具有降低的神经元或神经胶质空泡化的发生率和/或严重性。
非cns组织评估。当与mpsi或仅hidua对照(分别为轻度至中度和中度)相比时,给予zfn+hidua供体的动物中的主动脉空泡化(中膜)的严重性降低了。当与mpsi对照相比时,给予zfn+hidua供体的动物(第4和第5组)的肺、心脏瓣膜或间质、主动脉中膜、肾小管上皮、膀胱(尿道上皮和间质)、骨骼肌间质细胞、脾巨噬细胞/组织细胞、肠系膜淋巴结巨噬细胞/组织细胞、胸腺间质细胞、kupffer细胞、坐骨神经、甲状旁腺细胞、角膜基质/内皮细胞(descemet膜)、胃肠道(肌层和间质[雌性几乎不存在])、垂体远侧部和注射部位中的空泡化发生率和/或严重性降低了。在给予zfn+hidua供体(第5组)的雌性中,胸骨或股胫关节软骨细胞/骨细胞/骨膜细胞、股胫关节滑膜中的空泡化,以及卵巢、子宫、子宫颈和阴道中的空泡化的发生率降低了。在给予zfn+hidua供体的雄性中,存在附睾上皮或间质细胞的空泡化的发生率降低。
神经行为评价:巴恩斯迷宫。使用巴恩斯迷宫测试在尸检前的最后一周测试小鼠的认知表现。mpsi小鼠先前已在巴恩斯迷宫(belur等(2015).5月13日至16日在americansocietyofgeneandcelltherapyasgct上发表;neworleans,la)和其它认知能力的测试,诸如水t-迷宫(ou等,同上)两者中显示出学习缺陷。
巴恩斯迷宫测试发生在一个圆形平台上,其中四十个孔围绕平台中心环绕。参见,图6。所有孔都被阻塞,除了一个孔具有小鼠可从所述孔进入的“逃生箱”。明亮的顶部照明产生厌恶刺激,从而鼓励动物寻找目标逃生孔并避开光线。容易看见的视觉提示被放置在迷宫周围的墙壁上,并作为空间线索来定位小鼠(参见图6)。将小鼠在巴恩斯迷宫中训练6天,每天4次试验,其中每次试验的最大时限为3分钟。每只动物的连续试验之间的间隔为12-15分钟。
如图7a和7b分别对于雄性和雌性动物所示,如通过在试验的6天内用于发现目标逃生孔的时延的减少所指出的,野生型雄性小鼠随时间推移而改善,而未治疗的mpsi雄性小鼠的逃生时延随时间推移没有改善(在第4-6天,p<0.05相对于野生型)。相比之下,与制剂缓冲液治疗的mpsi组相比,接受替代小鼠zfn+hidua供体的雄性mpsi小鼠显示出迷宫表现随时间推移的改善(在第4-6天,p<0.05),并且与野生型动物表现同样好。到测试的第4天,zfn+供体治疗的雄性mpsi小鼠在学习行为方面显示出统计学上显著的差异,其中第6天发现目标孔的平均值为53±26(平均值±sem),而未治疗的雄性mpsi小鼠花费149±17秒才发现目标。野生型雄性小鼠在第6天在51±15秒内到达目标。
与野生型小鼠相比,接受制剂缓冲液的雌性mpsi小鼠在6个测试日内的表现没有显示出显著差异。然而,用zfn+供体治疗的雌性mpsi小鼠在研究过程中显示出更好的迷宫表现的趋势。在第6天,治疗的雌性mpsi小鼠花费38±23秒来发现目标孔,并且配制缓冲液处理的雌性mpsi小鼠花费96±33秒来发现目标。这些结果表明,hidua可能在mpsi小鼠中获得进入cns并产生积极神经行为作用而不需要修饰hidua蛋白以有利于穿过血脑屏障。(参见例如,ou等,同上)。因此,本文实现的一致的高idua血浆水平(由于肝脏的不断分泌)导致未修饰的idua穿过血脑屏障。
重要的是,gag数据指示在zfn+供体治疗的mpsi小鼠中,表达的idua活性与尿gag水平的正常化以及肝脏和二级组织中gag水平的降低相关。此外,所述治疗与cns和其它靶组织中空泡化减少的趋势以及巴恩斯迷宫测试中认知表现的改善(改善的空间记忆)相关。
总之,本文提供的数据表明,在mpsi受试者的白蛋白基因座处的zfn介导的基因替代治疗所述疾病。这项4个月的药理学/毒理学研究表明:通过肝脏向性raav2/8载体共同递送替代小鼠zfn与hidua转基因供体导致1)mpsi小鼠肝细胞中白蛋白基因座的体内有效修饰;2)肝脏中的高水平hidua酶表达和活性;3)功能性酶分泌到血浆中;4)由二级组织(包括脾脏、肺、心脏、肌肉和脑)从血浆摄取idua。此外,结果还表明,所实现的idua活性水平足以至少部分校正非靶组织中的代谢缺陷(gag积聚),这导致改善的行为功能。
实施例3:在小鼠模型中治疗mpsii(ids敲除或idsy/-)
与以上使用idua转基因治疗mpsi的工作类似,使用校正性人ids转基因在mpsii小鼠模型系统中重复实验。制备包含ids转基因的aav2/8病毒,连同靶向小鼠白蛋白基因的内含子1中的同源位点的小鼠zfnraav载体(sb-48641、sb-31523,显示在美国专利申请14/872,537中)以及编码侧接与小鼠基因座具有同源性的臂的无启动子人ids转基因(hids)的供体(sb-mu-ids)并用于治疗mpsii小鼠。本研究中使用的zfn:zfn:供体的比率为1:1:8。对于此小鼠研究,使用血清型raav2/8包装和递送替代试剂,因为其在小鼠体内赋予比raav2/6载体更好的转导和更快的转基因表达。将raav2/8载体稀释到制剂缓冲液,补充有35mmnacl和5%甘油,ph7.1的磷酸盐缓冲盐水(pbs)中。
研究设计如下表5所示,其列出了6组小鼠和所使用的每种类型aav2/8病毒的量。
表5:mpsii研究设计
此研究基本上如以上针对mpsi治疗所述地进行。如上所述进行蛋白质印迹,并使用针对人ids蛋白产生的抗体检测表达的人ids蛋白,以避免检测到小鼠蛋白(来自r&d系统的af2449)。结果显示,在1个月和4个月时接受zfn和供体两者的治疗组(上述第3、第4和第5组)中可检测到人ids蛋白。
通过溶解待分析的组织随后检测ids活性来检测ids活性。遵循的方法如下:
组织溶解:通过将小鼠组织样品与盐水(0.9%氯化钠)混合来制备组织蛋白提取物。将样品储存在冰上,并使用bullet
ids活性测定:使用合成底物4-甲基伞形酮基α-l-吡喃艾杜糖醛酸2-硫酸盐(4muids;santacruzbiotechnology)在荧光测定法中评价ids酶活性。
这是两步测定,由voznyi等描述((2001),j.inheritmetabdis24(6):675-80)。在第一步中,将4muids与测试样品一起孵育;如果存在ids,则其催化从底物去除硫酸盐基团。所得化合物,4-甲基伞形酮基αl艾杜糖醛酸苷(4mu艾杜糖醛酸苷),是第二种酶αl艾杜糖醛酸酶(idua)的底物。在第一步已孵育一段固定时间后,淬灭ids活性并加入过量的重组idua,以驱动4mu艾杜糖醛酸苷的裂解,从而产生荧光化合物4甲基伞形酮(4mu)。简而言之,将10μl稀释样品与20μl溶解在底物缓冲液(0.1m乙酸钠,ph5.0,10mm乙酸铅(ii))中的1.25mm4muids混合。然后将反应在37℃下孵育。
为了停止ids活性并将任何脱硫酸底物转化成4mu,向每个样品中加入20μl2x浓缩mcilvaine缓冲液(0.2m柠檬酸盐,0.4m磷酸盐,ph4.5),随后加入10μl的1μg/ml重组idua。将idua反应在37℃下孵育过夜,并且然后通过加入200μl终止溶液(0.5mnahco3/na2co3,ph10.7,0.2%tritonx100)终止。通过synergytmmx酶标仪(biotek,winooski,vt)上的荧光(ex365/em450)测量测定4mu的释放。
使用4mu(sigmaaldrich,st.louismo)产生标准曲线。绘制所得数据,将对数对数曲线拟合至数据,并使用此最佳拟合曲线计算测试样品中4mu的浓度。将酶活性表示为每小时测定孵育时间(仅第一反应步骤)、每ml血浆或每mg蛋白质溶解物释放的nmol4mu(nmol/小时/ml或nmol/小时/mg)。
在治疗后1个月和4个月时测量动物肝脏中的ids蛋白(图8a)。数据表明,在接受供体和zfn两者的所有组中都可检测到所述蛋白质。另外,在治疗组的血浆中随时间推移测量活性,并且在接受zfn和供体的组的所有样品中检测活性。在高剂量组检测到最高活性(图8b)。在4个月时的处死时间点,在来自接受zfn和供体的动物的所有组织中也检测到ids活性。还在研究中的动物的许多器官中测量活性(图8c),并且在所有测试组织(包括脑)中,与未治疗的mpsii小鼠相比,高剂量治疗组显示出显著更多的ids活性。
如实施例2中所述地测量组织gag水平,并且结果显示从高剂量动物分析的所有组织(除了脑以外)中的gag显著降低(图9a和9b)。
在治疗后4个月使用巴恩斯迷宫测试测试治疗组的认知表现(图10)。数据表示在6天的测试(每天平均4次试验)中动物发现目标所花费的时间的平均值±sem。每只个体小鼠的数据如下表6所示(表示为每个测试日每只小鼠到达孔的时间,以秒为单位)。个体小鼠的数据在水平方向上示出在表格中(其中每个小鼠编号为1至10,并且每个数据点是每个试验日完成的四次运行的平均值)并且每次试验(试验日1-6)垂直示出。在高治疗剂量下,接受治疗的那些动物与未治疗的mpsii动物之间存在显著差异。
表6:巴恩斯迷宫中mpsii治疗组的认知表现(秒)
如图所示,在高治疗剂量下,接受治疗的那些动物与mpsii动物之间存在显著差异。
实施例4:表征人细胞中产生的ids和idua蛋白
a.hepg2细胞(池)和hepg2亚克隆的产生和表征
在转导后3天通过miseq分析评价基本上如wo2015/089077中所述用halbzfnssbs#47171和sbs#47931转导的hepg2细胞和ids或idua供体的整合,并显示白蛋白基因座处59.27%插入缺失(hids)和62.27%插入缺失(hidua)。基于这种高水平的修饰,选择这些细胞进行亚克隆,并且然后将四个hids亚克隆(编号4、8、15和55)和三个hidua亚克隆(编号21、25和30)接种于24孔板中,并且通过ids或idua酶活性测定和elisa两者分析上清液的hids或hidua分泌(图11)。将仅用sb47171和sb47931zfn载体转导的hepg2细胞用作阴性对照(图11中的“仅zfn”)。
四个ids亚克隆显示功能性ids的分泌,其中这些亚克隆的上清液中的活性水平在408至1,855nmol/小时/ml范围内。所有亚克隆显示出比原始hepg2池高几倍的hids活性(图11a和11b中的‘ids-池’)。
三个idua亚克隆显示功能性idua的分泌,其中这些亚克隆的上清液中的活性水平在92-475nmol/小时/ml范围内(图11c和11d)。
这些结果通过hids或hiduaelisa证实。对于hids,所有四个亚克隆还显示hids分泌到细胞培养上清液中,其水平范围在156至438ng/ml之间,比原始hepg2池高几倍。在两种测定中,在仅用zfn转导的hepg2池中未看到背景,这表明hepg2细胞未以这些测定可检测的水平分泌内源性hids。类似地,对于hidua,所有三个亚克隆也显示hidua分泌到细胞培养上清液中,其水平范围在40.6-209.8ng/ml之间。在两种测定中,在未治疗hepg2细胞或仅用zfn转导的hepg2亚克隆中未看到背景,这表明hepg2细胞未以这些测定可检测的水平分泌内源性hidua。
b.通过
关于基因组白蛋白基因座处的修饰进一步表征hepg2亚克隆4、8、15和55。首先,为了确定ids(“sb-ids”)供体通过nhej或hdr整合的模式,使用特异于各整合方法的引物和探针在转基因5'末端处进行
结果显示,所有四种分析的zfn+供体治疗的亚克隆都显示nhej或hdr特异性试剂的信号,这证实了这些亚克隆中白蛋白基因座处sb-ids转基因的5'整合事件。亚克隆15和55的结果清楚地表明hdr是sb-ids整合的机制(图12c)。在所有三个
c.通过
关于基因组白蛋白基因座处的修饰进一步表征hepg2idua亚克隆21、25和30。首先,为了确定idua(“sb-idua”)供体通过nhej或hdr整合的模式,使用特异于各整合方法的引物和探针在转基因5'末端处进行
结果显示,所有三种分析的zfn+供体治疗的亚克隆都显示nhej或hdr特异性试剂的信号,这证实了这些亚克隆中白蛋白基因座处sb-idua转基因的5'整合事件。虽然亚克隆21的结果清楚地表明了通过hdr的hidua整合,但亚克隆25和30的整合机制尚不清楚。因此,对所有三个亚克隆进行了替代的基因分型方法。对于此基因分型,使来自亚克隆21、25和30的基因组dna经受放射性标记的pcr测定,以评价白蛋白基因座内zfn靶位点处5'转基因整合事件的存在(图12h)。所得凝胶证实克隆21的hdr整合、克隆25的nhej和克隆30的nhej(图12i)。箭头指向通过nhej(上箭头)或hdr(下箭头)插入特征的带区域。
d.表征在hepg2ids亚克隆中由白蛋白基因座产生的hids多肽
ids在肝脏中的体内表达以及然后靶组织的亚序列摄取取决于ids蛋白上的糖基化模式(图13)。为了进一步表征由白蛋白基因座产生的hids蛋白,对由hepg2ids和对照hepg2克隆产生的上清液进行蛋白质印迹。将两组表达hids的hepg2细胞用于此分析。第一组是高ids表达的混合克隆,其产生方式与以上分析的hepg2ids克隆相同。白蛋白基因座处的miseq分析显示此‘混合克隆’由69.9%未修饰的hepg2细胞、21.4%在非ids插入白蛋白等位基因中含有5bp缺失的hepg2ids亚克隆和一起占总群的6.8%的含有小缺失的三个其它亚克隆(图14中标记为“混合”)组成。
使用的第二hepg2ids克隆是克隆#55,一种以下表征的低表达ids克隆。将hepg2idua克隆用作阴性对照,因为它以与这些hepg2ids克隆相同的方式产生,但其中idua(mpsi中缺乏的基因)整合在白蛋白而不是ids处(图14中的‘对照’)。
人ids最初内源性表达为550个氨基酸的未成熟前体形式,含有信号肽和前肽两者。25个氨基酸信号肽和8个氨基酸前肽两者在翻译后修饰和hids蛋白成熟过程中被裂解(wilson等,(1990)procnatlacadsciusa.87(21)85315)。得到的73-78kda多肽在高尔基体中被磷酸化并糖基化成90kda前体,其随后在溶酶体中裂解成55kda中间体,并且最后是45kda成熟hids蛋白,其中释放18kda多肽(froissart等,(1995)biochem.j.309(pt2):425-30)。
为了确定这些hids形式中的哪一种在hids整合后从白蛋白基因座产生,对hepg2细胞沉淀、hepg2细胞培养上清液(由细胞分泌hids后)和用此hepg2细胞培养上清液孵育后未修饰的原代肝细胞沉淀(模拟二级组织的摄取)进行蛋白质印迹。
如图14所示,在由白蛋白产生hids的hepg2沉淀(‘hepg2沉淀')中发现了全长ids以及所有中间体和成熟多肽形式。ids的全长形式被视为成片条带,可能是由于这些生产细胞中新合成的ids的持续翻译后修饰(糖基化)。这表明,类似于天然存在的多肽,由肝癌hepg2细胞中的白蛋白基因座产生的hids蛋白经翻译后修饰和分泌。产生的酶在约90kda的迁移与在成纤维细胞(froissart等,同上)和cho细胞中产生的重组ids(
然后,对来自这些hepg2亚克隆的条件上清液的分析指示,全长hids是从所述细胞分泌的ids的主要形式,其中还仅检测到18kda条带的非常弱的条带(‘hepg2上清液')。当此条件细胞培养上清液与原始未修饰的原代肝细胞一起孵育时,这些肝细胞有效地从上清液摄取hids(‘原代肝细胞沉淀')。尽管在这些沉淀中可检测到全长hids,但很明显hids也能够易于被加工成蛋白质的成熟形式,因为55、45和18kda多肽都存在于这些沉淀中。最后,从上清液摄取的hids是活性的,因为与来自分泌hepg2细胞的hids的上清液一起孵育的原代肝细胞中hids活性增加6.510.3倍。
结合起来,这些数据表明由白蛋白产生的hids通常在原始细胞中加工成其成熟形式,由所述细胞分泌全长形式,并且这种全长hids被二级细胞摄取并有效加工成其成熟和活性形式。
e.表征由hepg2亚克隆中白蛋白基因座产生的hids和hidua的糖基化
已有人建议/证明,对于在几种细胞类型如肝细胞、神经元和神经胶质细胞中有效的细胞摄取和适当的溶酶体靶向,对ids或idua酶进行甘露糖6磷酸修饰是必需的(daniele等,(2002)biochimbiophysacta.1588(3):203-9)。因此,为了表征由白蛋白基因座产生和分泌的hids和hidua的糖基化模式,对hepg2ids或idua亚克隆上清液进行蛋白质印迹。ids含有8个潜在的n糖基化位点,其由高甘露糖、杂合和复合寡糖链的组合占据(froissart等,同上)。这些类型的糖基化可使用特定的糖苷酶来区分:肽-n-糖苷酶f(png酶f)能够从糖蛋白裂解所有3种类型的寡糖,而内切糖苷酶h(endoh)仅能够从n-连接的糖蛋白裂解高甘露糖和一些杂合寡糖,但不能裂解复合寡糖(图15a)。
为了确定在由白蛋白基因座产生和分泌的hids以及从其中idua在白蛋白处整合的hepg2亚克隆收获的对照上清液(对照)中存在哪种糖基化物种,从两个不同的hepg2ids群(混合和克隆55;如上所述)收获细胞培养上清液。使上清液保持未消化(u)或经受用png酶f(p)或endoh(e)进行的消化。图15b显示来自hepg2ids群的上清液的png酶f消化(p)导致完全改变至大约60kda的单一条带,这表明从蛋白质中去除了所有寡糖链(=非糖基化的hids)。相比之下,endoh消化(e)导致中间迁移率的成片条带。对照上清液未显示任何hids表达,这表明从未处理的细胞分泌很少甚至没有hids。
类似地,当对来自hepg2沉淀的蛋白质溶解物进行相同组的糖苷酶消化时(图15c),所见的三种主要的大ids形式(全长约73-78kda;中等约55kda;和成熟约45kda)都显示出明显分子量的相同改变。全长和中间条带两者都显示png酶f消化(p)的完全改变和endoh消化(e)的部分改变。成熟的45kda条带显示出同样组的改变,但在png酶f消化后更清晰可见,这与已发表的文献一致(froissart等,同上)。
将hepg2-idua克隆类似地用于分析所产生的idua的糖基化模式。图15d显示png酶f和endoh在hepg2-idua细胞上清液中产生与可商购的idua酶
结合起来,这些数据表明由白蛋白基因座产生和分泌的hids和idua蛋白含有高甘露糖/杂合糖基化(endo-h敏感)和复合糖基化(endo-h不敏感/png酶f敏感)的混合物。
f.甘露糖-6磷酸盐对细胞ids摄取的竞争
通过在补充有10%fbs和1x青霉素-链霉素-谷氨酰胺(thermofisherscientific)的伊格尔氏最小必需培养基(eagle'sminimalessentialmedia)(mem)(corning)中培养对照hepg2/c3a(atcc,crl10741;‘hepg2’)细胞或hepg2-ids克隆#8或#15来制备条件培养基。将细胞培养72小时以使ids在细胞培养上清液中积聚,并且然后收集上清液、0.2μm过滤,并在-80℃下冷冻直至使用。
将100,000个未修饰的hepg2或k562细胞接种于24孔板中的每个孔,并在37℃、5%co2培养箱中生长24小时。然后在存在或不存在5mm甘露糖-6-磷酸(m6p;sigma-aldrich)的情况下将培养基更换为hepg2或hepg2-ids条件培养基(如上所述产生)。将细胞在条件培养基±5mmm6p中孵育6或24小时。将在条件培养基中孵育6小时的细胞转换回正常细胞生长培养基18小时,并在此24小时期间结束时收集来自所有条件的细胞沉淀。在去除细胞培养上清液后,在测定ids活性之前,用1mlpbs洗涤细胞沉淀一次以去除任何残留的条件培养基。通过在150μltbs+0.1%tritonx-100(sigma-aldrich)和1xhalt蛋白酶抑制剂(thermofisherscientific)中超声处理裂解细胞沉淀,并测定ids活性。
结果(图16a和16b)证明,在5mmm6p存在下,hepg2和k562细胞两者中ids摄取都降低了。
g.由体外白蛋白基因座产生的idua以甘露糖-6-磷酸依赖性方式被二级细胞摄取。图16d是说明用甘露糖-6-磷酸(m6p)处理k562细胞如何防止idua酶摄取的示意图。
通过在补充有10%fbs和1x青霉素-链霉素-谷氨酰胺(thermofisherscientific)的伊格尔氏最小必需培养基(mem)(corning)中培养对照hepg2/c3a(atcc,crl-10741;‘hepg2’)细胞或hepg2-idua克隆#25来产生培养基。将细胞培养72小时以使idua在细胞培养上清液中积聚,并且然后收集上清液、0.2μm过滤,并在-80℃下冷冻直至使用。
如下测定idua摄取。在存在或不存在5mm甘露糖-6-磷酸(m6p;sigma-aldrich)的情况下,将100,000个未修饰的k562细胞接种于在hepg2条件培养基(如上所述产生)中的24孔板中的每个孔。将细胞在条件培养基±5mmm6p中孵育24小时。在去除细胞培养上清液后,用1mlpbs洗涤细胞沉淀两次以去除任何残留的条件培养基,然后使用荧光底物4-甲基伞形酮基α-l-艾杜糖醛酸苷(santacruzbiotechnology)测定idua活性。
如图16c所示,由体外白蛋白基因座产生的idua以甘露糖-6-磷酸依赖性方式被二级细胞摄取。
实施例5:供体摄入脑中
在pacificbiolabs(hercules,ca)进行脑组织匀浆的质谱分析以测定硫酸皮肤素和硫酸乙酰肝素的水平,其是作为idua和ids酶的底物的糖胺聚糖。简而言之,将如实施例2中所述mpsi小鼠(未治疗和用zfn和idua供体治疗)的组织在200mm乙酸铵、20mm乙酸钙、4mmdtt、ph7.0中进行均化。通过bca蛋白质测定试剂盒(thermofisherscientific)测定蛋白质浓度,并将样品标准化至1mg/ml。用肝素酶i和iii消化硫酸乙酰肝素样品,产生两种主要的二糖(δ-ua-glcnac[iv-a]和δ-ua-glcns[iv-s])。用软骨素酶b消化硫酸皮肤素样品,产生3种主要的二糖(δua-galnac(4s)[di-4s]、δua-galnac(6s)[δdi-6s]和δua-galnac(4s,6s)[δdi-4s,6s])。在消化后,掺入内标物,通过10,000nmwl旋转过滤器或滤板离心去除大分子,并使样品经受反相hplc。使用在负模式下的特定多反应监测方法通过esi-ms/ms检测分析物和内标物。将每个分析物和is的峰面积在它们各自的ms色谱图中积分,并且将峰面积比率用于定量。从已知浓度的标准物的校准曲线内插未知样品中各个二糖的浓度,并以μg/ml报告,其中每种二糖的定量下限(lloq)为0.005μg/ml。将每种样品的二糖浓度相加并相对于匀浆输入作图。
如图17a所示,当通过质谱法特异性地测量来自mpsi小鼠的脑组织匀浆的硫酸皮肤素和硫酸乙酰肝素水平时,在注射后4个月时与其性别匹配的对照相比,雄性zfn+供体治疗的小鼠中硫酸皮肤素水平显著降低了。雌性治疗的小鼠也显示出降低的硫酸皮肤素水平的趋势。同样,zfn+供体治疗的mpsii小鼠还显示出脑硫酸皮肤素水平降低的趋势,其在最高zfn+供体剂量下达到统计学显著性(图17b)。然而,在mpsi或mpsii小鼠模型中没有观察到硫酸乙酰肝素的降低。
所有本文提到的专利、专利申请以及专利公布据此全文以引用方式并入。
尽管出于清楚理解的目的,相当详细地以说明和实施例的方式提供了公开内容,但对于本领域技术人员将显而易见的是,在不脱离本公开的精神或范围的情况下可实践各种改变和修改。因此,前面的描述和实施例不应理解为具有限制性。
- 还没有人留言评论。精彩留言会获得点赞!