缀合物和缀合试剂的制作方法
本发明涉及新型缀合物和新型缀合试剂。
发明背景
近年来,许多研究致力于将多种有效负载,例如治疗、诊断和标记试剂缀合至肽和蛋白质以广泛应用。蛋白质或肽本身可具有治疗性质,且/或它可以为结合蛋白。
肽和蛋白质具有作为治疗剂的潜在用途,并且缀合是改善其性质的一种方法。例如,水溶性合成聚合物,特别是聚亚烷基二醇,广泛用于缀合具有治疗活性的肽或蛋白质。通过延长循环时间和降低清除率、降低全身毒性以及在一些情况下显示出提高的临床疗效,已显示这些治疗性缀合物有利地改变了药代动力学。通常将聚乙二醇peg共价缀合至蛋白质的方法称为“peg化”。peg链可以携带有效负载,例如治疗、诊断或标记试剂。虽然已经提出了peg的替代聚合物,但peg仍然是主要选择的聚合物。
结合蛋白,特别是抗体或抗体片段,经常被缀合。结合蛋白对靶细胞和分子表面上的特异性标记物的特异性使得它们本身广泛用作治疗或诊断试剂或者作为可包括治疗、诊断或标记试剂的有效负载的载体。此类与标记和报道基团(例如荧光团、放射性同位素和酶)缀合的蛋白质可用于标记和成像用途,而与诸如细胞毒性剂类和化疗药物类药物缀合以产生抗体-药物缀合物(adcs)则允许靶向递送此类药物至特定组织或结构,例如特定的细胞类型或生长因子,以减少对正常健康组织的影响并显著地降低与化疗相关的副作用。此类缀合物在多种疾病领域中,特别是在癌症中,都具有广泛的潜在治疗用途。
文献中已经报道了许多缀合蛋白质和肽的方法。可能最常用的方法涉及使用基于马来酰亚胺的缀合试剂。此类试剂描述于多篇出版物中,例如wo2004/060965。liberatore等人在bioconj.chem1990,1,36-50中以及delrosario等人在bioconj.chem.1990,1,51-59中描述了产生更均匀产物的替代方法,其描述了在蛋白质中,包括在抗体中,可通过二硫键用于交联的试剂的用途。wo2005/007197描述了一种将聚合物缀合至蛋白质的方法,其使用的新型缀合试剂能与源自蛋白质中的二硫键的两个硫原子缀合以产生新型硫醚缀合物,而wo2009/047500则描述了同一缀合试剂结合附着于蛋白质的多聚组氨酸标记的用途。wo2010/100430描述了能够通过蛋白质中的二硫键形成单个碳原子桥的试剂。其他与蛋白质缀合有关的文献包括wo2014/064423、wo2013/190292、wo2013/190272和ep2260873。
wo2014/064424描述了特定的adc,其中药物是美登素且抗体通过二硫键经交联而键合。wo2014/064423描述了特定的adc,其中药物是奥瑞斯他汀且抗体通过二硫键经交联而键合。这些文献的实施例中示出的接头含有peg部分,其中peg链的一端经由接头的其他部分连接至药物,而peg链的另一端经由接头的其他部分连接至抗体。这是adc的常见结构模式。
近年来,缀合物中将有效负载连接至蛋白质或肽的接头的重要性变得越来越明显。通常,要做的关键决定为究竟是需要具有可裂解的接头,即在给予缀合物时降解以释放游离的有效负载的接头,还是不可裂解的接头。另一个关键决定为在接头中是否包含peg。考虑到这些因素,原则上可以使用任意接头。然而,在实践中,接头结构的变化可导致缀合试剂或所得缀合物在性质上的差异。
环糊精是环状低聚糖,由5个以上葡萄糖(α-d-吡喃葡萄糖苷)单元结合成环(通常通过其1和4位连接)而构成。最常见的环糊精α-环糊精、β-环糊精和γ-环糊精,它们分别是6、7和8元环。环糊精的结构已经确定且可被描述为一端比另一端稍窄的螺旋状或筒状。该结构提供了非极性空腔,该空腔中可以存在较不亲水的分子,该分子通过环糊精的亲水壳保护免受外部环境的影响,这有助于将较不亲水的分子溶解在水中。在这类复合物中,客分子与环糊精的结合通常是非共价的且环糊精-药物复合物通常被称为“包合复合物”,其中环糊精“主”分子以非共价相互作用固定“客”药物分子。由于能够形成复合物,环糊精和环糊精衍生物已被广泛用作药物制剂中的辅料。
可选但不太常见地,环糊精已经与活性成分反应以形成共价缀合物。wo91/13100描述了共价附着于靶向载体的环糊精,所述靶向载体可以是抗体或其片段。wo90/02141公开了与诸如药物类试剂共价键合的环糊精。us2001/0034333讨论了使用单个单体环糊精的困难,并提出使用交联环糊精聚合物来解决这些问题。它描述了与交联环糊精聚合物共价连接的荧光有效负载,其后续使用nhs酯官能团缀合至抗体。
我们现已发现,将环糊精引入特定结构的缀合物中得到令人惊讶地有效的结果。在体外,这些缀合物的效果惊人。
发明概述
本发明提供了一种缀合物,通过接头将蛋白质或肽缀合至治疗、诊断或标记试剂,其中所述接头包含具有下述通式的蛋白质或肽键合部分:
其中pr代表所述蛋白质或肽,每个nu代表存在于蛋白质或肽中或附着于蛋白质或肽的亲核体,a和b各自独立地代表c1-4亚烷基或亚烯基链,并且w'代表吸电子基团或通过还原吸电子基团而得到的基团;并且其中所述接头还包括环糊精。
本发明还提供了一种缀合试剂,其能够与蛋白质或肽反应并包括治疗、诊断或标记试剂及接头,所述接头包括能够与蛋白质或肽反应的官能团,所述官能团为下式的基团:
其中w代表吸电子基团,a和b具有上述给定的含义,m为0至4,且每个l各自独立地代表离去基团;且其中所述接头也包括环糊精。
本发明还提供了一种制备本发明的缀合物的方法,其包括使蛋白质或肽与本发明的缀合试剂反应。
发明详述
本发明的缀合物包括通过接头与蛋白质或肽共价连接的治疗、诊断或标记试剂(有效负载),而本发明的试剂包括与式ii或ii'的官能团共价连接、能够与蛋白质或肽(蛋白质键合部分)反应的有效负载。本发明缀合物或试剂中的环糊精可以存在于接头的骨架内,或者其可以作为束缚在接头的骨架上的侧基而存在。具有前述结构的缀合物可以用式d~cd~f'示意性表示,其中d代表治疗、诊断或标记试剂,f'代表式i的基团,且cd代表环糊精,而具有相应结构的试剂可以用式d~cd~f示意性表示,其中d代表治疗、诊断或标记试剂,f代表式ii或ii'的基团,且cd代表环糊精。然而,本发明的缀合物和试剂优选具有后一种结构,即环糊精作为束缚在接头的骨架上的侧基而存在。具有该结构的本发明的缀合物可用下式示意性表示:
其中d代表治疗、诊断和标记试剂,f’代表式i的基团,且cd代表环糊精,而本发明的试剂可通过下式示意性表示:
其中d代表治疗、诊断和标记试剂,f代表式ii或ii'的基团,且cd代表环糊精。官能基f能够与存在于蛋白质或肽中的两个亲核体反应,如下解释。
环糊精
环糊精是α-d-吡喃葡萄糖苷单元的环状低聚物。环状葡萄糖单元在其1,4-位上结合在一起。各种大小的环都可能,最常见的是在环内具有6个糖类半分子的α-环糊精;在环内具有7个糖类半分子的β-环糊精;和在环内具有8个糖类半分子的γ-环糊精。这些环糊精是天然存在的,如下所示:
其他具有不同环大小的环糊精可通过已知方法合成制备或者酶催化制备,例如由endo&ueda,fabadj.pharm.sci.,2004,29,27-38所述。在本发明的一个实施方案中,环糊精是α-环糊精;在另一个实施方案中,环糊精是β-环糊精;在另一个实施方案中,环糊精是γ-环糊精。
环糊精可以是单环(即由单个环糊精环构成),或者可以存在形成环糊精二聚体或聚合物的两个以上的环糊精环。环糊精二聚体和聚合物是已知的,且可以通过已知方法合成。环糊精优选为单环。
天然环糊精中的每个环状葡萄糖单元携带三个羟基,其中两个在2和3位上并由葡萄糖环直接携带,而一个在6位上且是羟甲基的一部分。这些羟基中的一个或多个可以被任意其他所需基团替代以形成衍生化的环糊精。该衍生化的环糊精应理解为在本发明的范围内。例如,羟基可被卤原子替代;或者羟基可被式-yrb的基团替代,其中y代表-o-、-s-、-o-co-、-co-o-、-co-nrb-、-nrb-co-、-co-、-so-、-so2-、-s-co-、-co-s-、-n=crb-、-ch=n-rb、-o-co-o-、-o-so2-、-so2-o-或-o-so2-o-,且rb代表氢原子或烷基(优选c1-6烷基,例如甲基)、烯基(优选c2-6烯基)、炔基(优选c2-6炔基,例如炔丙基)、芳基(优选苯基)或烷基-芳基(优选c1-6烷基苯基)基团,这些基团各自未被取代的或被一个或多个羟基取代;或者羟基可以被式-rb、nrbrb、=nrb、-sirbrbrb、-o-sirbrbrb、prbrb、-po-rbrb或-o-po(orb)2的基团替代,其中每个rb独立地具有上述给定的含义。
可替代羟基的优选基团包括卤原子或氨基、巯基、叠氮化物、烷基、羟基烷基、羟基烯基、羟基炔基或烷基甲硅烷氧基。还优选羟基被酯化形成硫酸酯或磷酸酯的环糊精。许多衍生化的环糊精可市售获得,并且这些中的任何一种都可用于本发明中。例如,下述α-环糊精的衍生物可市售获得(一些以盐的形式):六(6-o-叔丁基二甲基甲硅烷基)-α-环糊精;六(6-叠氮基-6-脱氧)-α-环糊精;六(6-氨基-6-脱氧)-α-环糊精;六(6-溴-6-脱氧)-α-环糊精;六(6-碘-6-脱氧)-α-环糊精;六(6-疏基-6-脱氧)-α-环糊精;羧甲基-α-环糊精;α-环糊精磷酸盐;α-环糊精硫酸盐;(2-羟丙基)-α环糊精;六(3-氨基-3-脱氧)-α-环糊精;和甲基-α-环糊精。
下述β-环糊精的衍生物可市售获得:七(6-o-叔丁基二甲基甲硅烷基)-β-环糊精;七(6-叠氮基-6-脱氧)-β-环糊精;七(6-氨基-6-脱氧)-β-环糊精;七(6-溴-6-脱氧)-β-环糊精;七(6-脱氧-6-碘)-β-环糊精;6-单甲苯磺酰基-β-环糊精;6-单脱氧-6-单氨基-β-环糊精;七(6-脱氧-6-巯基)-β-环糊精;羧甲基-β-环糊精;β-环糊精磷酸盐;β-环糊精硫酸盐;(2-羟丙基)-β-环糊精;七(3-氨基-3-脱氧)-β-环糊精;a,d-6-二氨基-6-双脱氧-β-环糊精;和甲基-β-环糊精。
下述γ-环糊精的衍生物可市售获得:八(6-o-叔丁基二甲基甲硅烷基)-γ-环糊精;八(6-叠氮基-6-脱氧)-γ-环糊精;八(6-氨基-6-脱氧)-γ-环糊精;八(6-溴-6-脱氧)-γ-环糊精;八(6-脱氧-6-碘)-γ-环糊精;和八(6-脱氧-6-疏基)-γ-环糊精;羧甲基-γ环糊精;γ-环糊精磷酸盐;γ-环糊精硫酸盐;(2-羟丙基)-γ环糊精;八(3-氨基-3-脱氧)-γ-环糊精;和甲基-γ-环糊精。
这些的任何一种都可以引入本发明的缀合物或试剂中。
在本发明的优选实施方案中,存在于一个葡萄糖环中的3-羟基或6-羟基或两者被nh2基团替代。这提供了一种将环糊精与本发明的缀合物或试剂的接头共价键合的方便方法,如下所述。能够取代3-和/或6-羟基以提供方便合成途径的替代基团包括硫醇基、叠氮基、-o-炔丙基、醛基和羧基。
环糊精可从一个或多个环状葡萄糖基团中的任意合适位置被键合至接头的其他部位。在一个优选的实施方案中,环糊精通过3-或6-位与接头的其余部分键合。其他位点也可以提供键合位点,要么通过羟基,要么通过如上所述的取代基。
有效负载
本发明的缀合物和试剂携带有效负载,其是治疗、诊断或标记试剂。该有效负载通过接头与环糊精并且与蛋白质或肽共价键合。可存在单分子的治疗、诊断或标记试剂,或者可存在两个以上分子。优选包含一个或多个药物分子,例如细胞毒性剂或毒素。奥瑞斯他汀类(auristatins)、美登素类(maytansinoids)或多卡米星类(duocarmycins)是典型的细胞毒性药物。通常优选的是,药物缀合物,特别是抗体药物缀合物,应含有药物的多个拷贝。标记试剂(应理解为包括显像剂)可以例如包括放射性核素、荧光剂(例如胺衍生的荧光探针,例如5-二甲基氨基萘-1-(n-(2-氨基乙基))磺酰胺-丹酰基乙二胺、
优选地,有效负载为治疗试剂,尤其是上述之一。
已知治疗、诊断或显像试剂可与环糊精形成复合物,其中所述试剂通过非共价键合与环糊精复合。除了作为本发明的缀合物所需的基本特征,本发明的缀合物还可以含有第二治疗、诊断或显像试剂,特别是第二治疗试剂,所述第二试剂以与环糊精的复合物的形式存在。
蛋白质
为了方便,在本部分和其他部分中,除非上下文另有要求,“蛋白质”应理解为包括“蛋白质和肽”。
可存在于本发明的缀合物中的合适的蛋白质包括例如肽、多肽、抗体、抗体片段、酶、细胞因子、趋化因子、受体、血液因子、肽激素、毒素、转录蛋白或多聚体蛋白。
酶包括碳水化合物特异性酶、蛋白水解酶等,例如us4,179,337公开的氧化还原酶、转移酶、水解酶、裂解酶、异构酶和连接酶。引人注意的特异性酶包括天冬酰胺酶、精氨酸酶、腺苷脱氨酶、超氧化物歧化酶、过氧化氢酶、胰凝乳蛋白酶、脂肪酶、尿酸酶、胆红素氧化酶、葡萄糖氧化酶、葡糖醛酸糖苷酶、半乳糖苷酶、葡糖脑苷脂酶(glucocerbrosidase)和谷氨酰胺酶。
血液蛋白包括白蛋白、运铁蛋白、因子vii、因子viii或因子ix、血管假性血友病因子、胰岛素、acth、胰高血糖素、生长抑素、生长激素、胸腺素、甲状旁腺激素、色素激素、生长调节素、促红细胞生成素、黄体生成素、下丘脑释放因子、抗利尿素、催乳素、白细胞介素,干扰素例如ifn-α或ifn-β、集落刺激因子、血红蛋白、细胞因子、抗体、抗体片段、绒毛膜促性腺激素、促卵泡激素、促甲状腺激素和组织纤维蛋白溶酶原激活剂。
其他引人注意的蛋白质为由dreborg等人在crit.rev.therap.drugcarriersyst.(1990)6315-365中公开的过敏原蛋白,由于与诸如聚(环氧烷)的聚合物缀合时具有降低的变应原性并因此适合用作耐受诱导剂。在所公开的过敏原中有豚草抗原e、蜜蜂毒液、螨过敏原等。
引人注意的是糖多肽如免疫球蛋白、卵清蛋白、脂肪酶、葡糖脑苷脂酶、凝集素、组织纤维蛋白溶酶原激活剂和糖基化白细胞介素、干扰素和集落刺激因子以及免疫球蛋白如igg、ige、igm、iga、igd及其片段。
特别引人注意的是受体和配体结合蛋白以及临床医学用于诊断和治疗目的的抗体和抗体片段。抗体-药物缀合物,特别是当药物是细胞毒类药物例如奥瑞斯他汀、美登素类或多卡米星时,是本发明特别优选的实施方案。除非上下文另有要求,否则本说明书中对本发明的缀合物的任何引用均应理解为包括对抗体药物缀合物的特定引用。
如有需要可以将蛋白质衍生化或官能化。特别是在缀合之前,蛋白质例如天然蛋白质可以先与各种阻断基团反应以保护其上的敏感基团;或者它可以先与一种或多种聚合物或其他分子缀合。它可以含有聚组氨酸标记,其在缀合反应期间可以被缀合试剂靶向。
蛋白质或肽的结合,及缀合试剂
本发明的缀合试剂为在wo2005/007197和wo2010/000393中公开的一般类型。官能团ii和ii'是彼此的化学等价物。当含有基团ii的试剂与蛋白质反应时,失去第一离去基团l以原位形成含有与第一亲核体反应的基团ii'的缀合试剂。然后失去第二离去基团l,并与第二亲核体反应。因此,作为将含有官能团ii的试剂用作起始材料的替代方案,含有官能团ii'的试剂可被用作起始材料。
离去基团l可为例如-sp、-op、-so2p、-oso2p、-n+pr2r3、卤素或
其中p代表包含-(ch2ch2o)n-部分的基团且其中n为2或更大的数值的缀合试剂是我们共同未决的申请gb1418186的主题,wo2016/059377要求其优先权。该申请公开了如下内容:
“离去基团可包括例如-(ch2ch2o)n-r1,其中r1为封端基团。可使用非常广泛的封端基团。r1可为例如氢原子、烷基(特别是c1-4烷基,特别是甲基)或任选取代的芳基(例如任选取代的苯基,例如甲苯基)。或者,封端基团可包括官能团例如羧基或氨基。这种封端基团可例如具有式-ch2ch2co2h或-ch2ch2nh2,且可通过将-(ch2ch2o)n-链的末端单元官能化来制备。或者,除被封端基团封端以外,-(ch2ch2o)n-基团在缀合试剂中可具有两个连接点,从而在化学上相当于存在两个能够与两个亲核体反应的离去基团。
离去基团的-(ch2ch2o)n-部分基于peg、聚乙二醇。peg可为直链或支链,其可以以任何方式被衍生化或官能化。n为2或更大的数值,例如2、3、4、5、6、7、8、9或10。例如,n可为5至9。或者,n可为10或更大的数值。对于n没有特定的上限。n可为例如150或更小,例如120或更小,例如100或更小。例如n可为2至150,例如7至150,例如7至120。离去基团的peg部分-(ch2ch2o)n-的分子量可为例如1至5kda;其可为例如1kda、2kda、3kda、4kda或5kda。如果需要,离去基团可含有两个或更多个被一个或更多个间隔基分隔的-(ch2ch2o)n-部分。
在本发明的缀合试剂中的离去基团适合地具有式-sp、-op、-so2p、-oso2p、-n+pr2r3,其中p为包括-(ch2ch2o)n-部分的基团,且r2和r3各自独立地代表氢原子、c1-4烷基,或基团p。优选地,r2和r3各自代表c1-4烷基(特别是甲基),或特别是氢原子。或者,缀合试剂可包括式-s-p-s-、-o-p-o-、-so2-p-so2-、-oso2-p-oso2-和-n+r2r3-p-n+r2r3-的基团。该类型的具体基团包括-s-(ch2ch2o)n-s-、-o-(ch2ch2o)n-o-、-so2-(ch2ch2o)n-so2-、-oso2-(ch2ch2o)n-oso2-或–n+r2r3-(ch2ch2o)n-n+r2r3-。它们还可包括以下类型的基团:
其中-(ch2ch2o)n-基团由任何合适的连接基团例如烷基携带。这些二价基团在化学上相当于两个能够与两个亲核体反应的离去基团。”
存在于本发明的新型缀合试剂中的特别优选的离去基团l为–sp或-so2p,特别是-so2p。在该基团中,一个优选的实施方案是其中p代表苯基,或特别是甲苯磺酰基。另一优选的实施方案是其中p代表包括-(ch2ch2o)n-部分的基团,特别是其中n具有一个上述值,尤其是7的基团。特别优选的离去基团l是-so2-(ch2ch2o)n-h/me,特别是-so2-(ch2ch2o)7-h/me。在本说明书全文中,任何对离去基团l的提及应被理解为包括对这些优选基团的具体提及,特别是-so2-(ch2ch2o)n-h/me,尤其特别是-so2-(ch2ch2o)7-h/me。
吸电子基团w可为例如酮基-co-、酯基-o-co-或砜基-so2-。优选地,w'代表这些基团中的一个或可由这些基团中的一个还原得到的基团,如下文所述。优选地,w代表酮基,优选地,w'代表酮基或可由酮基还原得到的基团,特别是ch.oh基团。
优选地基团f'和f具有下式:
特别是
蛋白质中的亲核体由例如半胱氨酸、赖氨酸或组氨酸残基提供,nu可为例如硫原子或氨基。在本发明的一个优选的实施方案中,每个nu代表存在于蛋白质的半胱氨酸残基中的硫原子。这种结构可以由存在于蛋白质中的二硫键的还原而得到。在另一个实施方案中,每个nu代表与所述蛋白质连接的聚组氨酸标记中的组氨酸残基中存在的咪唑基团。
接头
将治疗、诊断或标记试剂连接至本发明的缀合物中的蛋白质或肽键合部分或连接至本发明的缀合试剂中的官能团的接头必须包括一个或更多个如上所述的环糊精。其还可以含有其他任何所需的基团,特别是在该领域常见的任何常规基团。
小节(i)。在一个实施方案中,位于有效负载和式f'/f的基团之间的接头,特别是紧邻式f'/f的基团的接头部分,可包括亚烷基(优选c1-10亚烷基),或任选取代的芳基或杂芳基,其中的任何一个可以被一个或多个氧原子、硫原子、-nra基团(其中ra代表氢原子或烷基(优选c1-6烷基)、芳基(优选苯基),或烷基-芳基(优选c1-6烷基-苯基)基团)、酮基、-o-co-基团、-co-o-基团、-o-co-o、-o-co-nra-、-nr-co-o-、-co-nra-和/或-nra.co-基团终止或中断。合适的芳基包括苯基和萘基,而合适的杂芳基包括吡啶、吡咯、呋喃、吡喃、咪唑、吡唑、噁唑、哒嗪、嘧啶和嘌呤。紧邻f/f'基团的接头部分特别优选芳基或杂芳基,尤其是苯基。
芳基或杂芳基可以与连接基的另一部分相邻,所述连接基为或含有-nra.co-或-co.nra基团,例如-nh.co或-co.nh-基团。此处和本说明书中出现基团ra的其他地方,ra优选为c1-4烷基,尤其为甲基,或尤其为氢原子。
可存在于任选取代的芳基(特别是苯基)或杂芳基上的取代基包括例如一个或多个相同或不同的选自以下的取代基:烷基(优选c1-4烷基,特别是甲基,任选地被oh或co2h取代)、-cn、-cf3、-no2、-nra2、-co2ra、-coh、-ch2oh、-cora、-ora、-ocora、-oco2ra、-sra、-sora、-so2ra、-nracora、-nra.co2ra、-no、-nra.oh、-ch=n-nra.cora、-n+ra3、卤素例如氟或氯、-c≡cra和-c=cra2,其中每个ra独立地代表氢原子或烷基(优选c1-6烷基)、芳基(优选苯基),或烷基-芳基(优选c1-6烷基-苯基)基团。特别优选存在吸电子取代基。优选的取代基包括例如cn、no2、-ora、-ocora、-sra、-sra、-nra.cora、-nhoh和-nra.co2ra,特别是cn和no2。
优选地,接头包括与基团f'/f相邻的上述基团中的一个。特别优选包括以下基团的缀合物和缀合试剂:
或特别是:
任何上述结构都可以与下述小节(ii)和(iii)中提到的任何结构相邻。
在所有的上式iii、iv、v和vi中,优选地,f'具有上式i例如ia或ib,且优选地,f具有上式ii或ii',例如iia、iib、ii'a或ii'b。
小节(ii)。在一个实施方案中,接头可含有可降解基团,即其可含有在生理条件下断裂的基团,将有效负载与其键合或待键合的蛋白质分离。或者,其可为在生理条件下不裂解的接头。当接头在生理条件下断裂时,优选在细胞内条件下裂解。当靶标在细胞内时,优选接头对细胞外条件基本不敏感(即,从而不会阻止向细胞内靶标递送足够剂量的治疗试剂)。
当接头含有可降解基团时,其通常对水解条件敏感,例如其可为在某些ph值(例如酸性条件)下降解的基团。水解/酸性条件可见于例如核内体或溶酶体中。在酸性条件下易于水解的基团的实例包括腙类、缩氨基脲类、缩氨硫脲类、顺式乌头酰胺类(cis-acotinicamides)、原酸酯类和缩酮类。易受水解条件影响的基团的实例包括:
在一个优选的实施方案中,接头包括
例如,接头可包括:
所述接头还可在还原条件下易于降解。例如,其可含有一旦暴露于生物还原剂例如硫醇下就可裂解的二硫基团。二硫基团的实例包括:
其中r、r′、r″和r″′各自独立地为氢或c1-4烷基。在一个优选的实施方案中,接头包括
例如,接头可包括
接头还可含有易于酶解的基团,其例如可易于被蛋白酶(例如溶酶体或核内体蛋白酶)或肽酶裂解。在本发明特别优选的实施方案中,接头的一部分含有肽基,所述肽基含有至少一个,例如至少两个、至少三个、至少四个或至少五个氨基酸残基,特别是天然存在的α氨基酸。例如,接头的该部分可含有序列phe-leu、gly-phe-leu-gly、val-ala、val-cit、phe-lys或glu-glu-glu,且优选存在val-cit肽基团。如下所述,特别优选含有序列val-cit-pab的接头。
易于酶解的基团的特别优选的实例是:
其中aa代表氨基酸序列,尤其是蛋白酶特异性氨基酸序列,例如上述之一,特别是val-cit。
在一个优选的实施方案中,接头包括:
例如,接头可包括
所述接头可携带单个有效负载d,或多于一个基团d。通过使用支化接头可引入多个基团d,例如其可引入天冬氨酸或谷氨酸或类似的残基。这引入下式的分支元件:
其中b为1、2、3或4,b=1为天冬氨酸、b=2为谷氨酸、且b=3代表一个优选的实施方案。在上式中的每个酰基部分可与基团d偶联。上述分支基团可引入-co.ch2-基团,因此:
如果需要,天冬氨酸或谷氨酸或类似的残基可与其他天冬氨酸和/或谷氨酸和/或类似的残基偶联,例如:
等。
以类似的方式,可以引入氨基酸赖氨酸、丝氨酸、苏氨酸,半胱氨酸、精氨酸或酪氨酸或类似残基以形成分支基团,因此:
其中,对于赖氨酸而言b为4,且
其中,对于丝氨酸而言b为1。
可以使用类似的分支基团将环糊精基团引入接头中,并且这样的结构形成本发明的另一个优选实施方案。因此,例如上述分支元件之一,例如天冬氨酸、谷氨酸、赖氨酸或丝氨酸或类似残基可以与一个分支一起存在,从而产生治疗、诊断或标记试剂例如药物d,而其他则产生含有环糊精基团的分支。上述各种接头部分可以存在于分支基团前或后的任何位置。
显而易见的是,f/f'基团和有效负载之间的许多接头的替代构型是可能的。一种优选构型可以示意性地表示如下:
其中,e代表以上小节(i)中所述的基团之一,且x代表本小节(ii)中所述的基团之一。
一个具体的特别优选的结构如下所示:
其中d、cd、f'和f具有如上给定的含义。方便地,环糊精可通过酰胺键键合至上式中的接头的其余部分,因此:
这样的结构尤其优选的实例如下:
这样的缀合物和试剂可以由其中3-或6-羟基已经被nh2基团替代的环糊精制备,如上所述。
小节(iii)。如果需要,将治疗、诊断或标记试剂连接至本发明的缀合物中的蛋白质或肽或连接至本发明的缀合试剂中的官能团上的接头可含有peg,或不含有peg。例如,其可在接头的骨架中含有peg,因此示意性地表示为:
可选地或另外地,peg可以作为接头上的侧基存在,示意性地表示为:
在这些式中,p、q和r代表存在于缀合物或试剂的接头中的各种可能的peg链中存在的乙二醇单元的数目。为了清晰起见,peg单元显示为直链单元,但应理解为所述单元中的任何一个可包括支链。
如果存在peg,本发明中的缀合物和试剂中存在的~(ch2-ch2-o-)~单元的总数量当然将取决于预期应用。在一些应用中,可使用高分子量peg,例如数均分子量可高达约75,000,例如高达50,000、40,000或30,000g/mol。例如,数均分子量可在500g/mol至约75,000的范围内。但是就一些应用而言,优选较小的peg部分。例如peg部分的分子量可高达3,000g/mol。尽管如此,对于一些应用而言,含有少至2个乙二醇重复单元,例如2至50个重复单元的peg基团是有用的。例如可使用具有2、3、4、5、6、7、8、9或10个重复单元或12、20、24、36、40或48个重复单元的含peg部分。
小节(iv)。将治疗、诊断或标记试剂连接至本发明的缀合物中的蛋白质或肽或连接至本发明的缀合试剂中的官能团上的接头可含有两个或更多个环糊精,且这些环糊精可以存在于接头的骨架内或作为束缚在接头的骨架上的侧基。因此,对于两个侧基环糊精,这可以示意性地表示:
且很明显可以同样存在多于两个这样的基团。
可以使用任何合适的方法向接头中引入多个环糊精。例如通过与存在于上述任何接头部分中的任何反应性基团反应可引入多肽链。可使用上述式的分支基团。例如,在一个具体的实施方案中,通过使用下述结构可引入两个环糊精:
或者,可通过使用多元醇官能度引入分支,例如:
~chs[(ch2)to~]3-s~
其中s为0、1或2,且t为1至4。例如,在一个具体的实施方案中,可通过使用以下结构引入三个侧基多肽链:
~c[ch2o-(ch2)2-co-nh~cd]3
缀合方法
本发明的缀合试剂可以与蛋白质或肽反应以形成本发明的缀合物,且该反应构成了本发明的另一方面。因此,包含官能团ii或ii'的缀合试剂与蛋白质或肽反应以形成包含基团i的缀合物。所述缀合方法的中间产物为含有吸电子基团w的缀合物。但是,所述缀合方法在合适的条件下是可逆的。这对于某些应用来说可能是期望的,例如需要快速释放蛋白质的应用,但对于其他应用,快速释放蛋白质可能是不期望的。因此,可能期望通过还原吸电子部分w以得到阻止蛋白质释放的部分来稳定缀合物。因此,上述方法可包括额外的任选步骤来还原缀合物中的吸电子基团w。使用硼氢化物,例如硼氢化钠、氰基硼氢化钠、硼氢化钾或三乙酰氧基硼氢化钠,作为还原剂是特别优选的。可使用的其他还原剂包括例如氯化锡(ii),醇盐例如烷醇铝,以及氢化铝锂。
因此,例如,含有酮基的部分w可被还原成含有ch(oh)基团的部分;醚基ch.ora可由羟基和醚化剂反应得到;酯基ch.o.c(o)ra可由羟基和酰化剂反应得到;氨基ch.nh2、ch.nhra或ch.nra2可由酮通过还原性胺化反应制得;或酰胺ch.nhc(o)ra或ch.n(c(o)ra)2可由胺的酰化形成。砜可被还原成亚砜、硫化物或硫醚。
使用本发明的缀合试剂的关键特征在于,α-亚甲基离去基团和双键与充当迈克尔活化部分的吸电子官能团交叉缀合。如果离去基团在交叉官能化试剂中易于消除而非直接置换,且吸电子基团为适合迈克尔反应的活化部分,则后续的分子内双烷基化可通过连续的迈克尔反应和逆-迈克尔反应进行。离去部分用于掩蔽潜在的缀合双键,其直到发生第一烷基化反应得到包含官能团ii'的试剂,以及由相继且交互式迈克尔反应和逆-迈克尔反应产生的双烷基化反应后才暴露。交叉官能性烷基化试剂可包含缀合至双键的多重键或位于离去基团和吸电子基团之间的多重键。
当与蛋白质的键合是经由来自蛋白质中二硫键的两个硫原子而进行时,所述方法可通过还原二硫键,随后还原产物与本发明的试剂反应来进行。可使用常规的方法,例如使用二硫苏糖醇、巯基乙醇或三羧基乙基膦,来还原二硫键。
可在与已知的缀合方法类似的条件下进行缀合反应,包括wo2005/007197、wo2009/047500、wo2014/064423和wo2014/064424中公开的条件。所述方法例如可在所有反应物均可溶的溶剂或溶剂混合物中进行。例如,蛋白质可以与聚合物缀合试剂在水性反应介质中直接反应。该反应介质也可以被缓冲,这取决于亲核体的ph要求。反应的最佳的ph一般为至少4.5,通常约5.0至约8.5,优选约6.0至7.5。最佳的反应条件当然将取决于所用的具体反应物。
当使用水性反应介质时,3-40℃的反应温度通常是合适的。在有机介质(例如thf、乙酸乙酯、丙酮)中进行的反应通常在最高达环境温度的温度下进行。在一个优选的实施方案中,所述反应在水性缓冲剂中进行,所述水性缓冲剂可包含部分有机溶剂,例如最高达20体积%的有机溶剂,通常5至20体积%的有机溶剂。
使用化学计量相当或稍微过量的缀合试剂可有效地缀合蛋白质。但是,还可使用化学计量过量的缀合试剂进行缀合反应,且这对于一些蛋白质而言是期望的。过量的试剂可例如通过离子交换色谱法或hplc在后续缀合物的纯化过程中容易地除去。
当然,当蛋白质含有足够多适合的连接点时,多于一个缀合试剂与蛋白质缀合是可能的。例如,在含有两个不同的二硫键的蛋白质中,或在含有一个二硫键并携带聚组氨酸标记的蛋白质中,每分子蛋白质可缀合两分子的试剂,并且这种缀合物构成本发明的一部分。
药物组合物和用途
其中有效负载是治疗试剂的本发明的缀合物可用于治疗各种医学疾病,这取决于有效负载的种类。通常,有效负载是细胞毒性剂且本发明可用于治疗癌症。因此,本发明进一步提供一种本发明的缀合物,特别是其中有效负载是治疗试剂的缀合物,且尤其是如下的缀合物:其为抗体-药物缀合物,连同药学上可接受的载体,及任选地连同其他活性成分。本发明进一步提供该种缀合物在治疗中的用途,并发现了在患者治疗方法中的用途,其包括向患者施用本发明的缀合物或药物组合物。本发明进一步提供本发明的缀合物在制备用于治疗例如癌症的药物中的用途。
附图说明
图1至图4示出了实施例11的结果。
以下实施例示例说明了本发明。
实施例1:含有氨基-6’-β-环糊精和奥瑞斯他汀细胞毒性有效负载mmae的缀合试剂1的合成。
步骤1:化合物2的合成。
向fmoc-glu-(oh)-oall(48mg)的dmf(1ml)溶液中加入hatu(110mg),并将溶液在0℃下搅拌30分钟。向其中加入
val-cit-pab-mmae.tfa盐(levenabiopharma、120mg)和nmm(32μl)的dmf(1ml)溶液。将反应混合物在室温下搅拌2.5小时。将溶剂真空浓缩并将粗产物溶于dmf(1.5ml)中,然后加入nmm(32μl)。将四(三苯基膦)钯(0)(45mg)加入反应混合物中,然后在室温下搅拌20小时。将反应溶液真空浓缩并通过反相c18柱色谱法用洗脱剂a(v/v):水:0.05%三氟乙酸和洗脱剂b(v/v):乙腈:0.05%三氟乙酸(100:0v/v至0:100v/v)洗脱而纯化残余物。真空除去有机溶剂并通过冷冻干燥除去含水溶剂以得到为白色固体的化合物2(98mg)。m/z[m+h]+(1475da,100%),[m+2h]2+(738,50%)。
步骤2:化合物3的合成。
向化合物2(23mg)的dmf(300μl)溶液中加入hatu(7mg),并将混合物在0℃下搅拌20分钟。将nmm(2μl)加入反应混合物中,将其在室温下再搅拌10分钟。将6-单脱氧-6-单氨基-β-环糊精盐酸盐(20mg)的dmf(100μl)溶液中加入nmm(2μl)并将溶液在室温下搅拌15分钟。然后将两种溶液合并并将额外量的hatu(7mg)和nmm(2μl)加入至合并的溶液中,将其在室温下搅拌2小时。然后添加哌啶(16μl)并在室温下搅拌反应混合物0.5小时。将反应溶液真空浓缩并将残余物通过反相c18柱色谱法使用洗脱液a(v/v):水:0.05%三氟乙酸和洗脱剂b(v/v):乙腈:0.05%三氟乙酸(100:0v/v至0:100v/v)洗脱进行纯化。真空除去有机溶剂并通过冷冻干燥除去含水溶剂以得到为白色固体的化合物3(15mg)。m/z[m+h]+(2369,15%),[m+2h]2+(1185,100%)。
步骤3:化合物4的合成。
向4-[2,2-双[(对甲苯磺酰)-甲基]乙酰基]苯甲酸(1.5g、natureprotocols,2006,1(54),2241-2252)于dmf(70ml)中的搅拌溶液中加入α-甲氧基-ω-巯基七(乙二醇)(3.2g)和三乙胺(2.5ml)。在室温下,在惰性氮气氛下搅拌所得反应混合物。19小时后,真空除去挥发物。将所得残余物溶解于水(2.4ml)中并通过反相c18柱色谱法使用洗脱液a(v/v):水:5%乙腈:0.05%三氟乙酸和洗脱剂b(v/v):乙腈:0.05%三氟乙酸(100:0v/v至0:100v/v)洗脱进行纯化。真空除去有机溶剂并通过冷冻干燥除去含水溶剂以得到为浓稠透明无色油状物的化合物4(1.8g)。m/z[m+h]+901。
步骤4:试剂5的合成。
在室温下向4(1.32g)的甲醇:水(18ml,9:1v/v)的搅拌溶液中加入
步骤5:试剂1的合成。
向试剂5(6.3mg)的dmf(250μl)溶液中加入hatu(2.6mg)并将溶液在0℃下搅拌20分钟。将nmm(0.5μl)加入至溶液中,并将溶液在室温下再搅拌10分钟。将nmm(0.75μl)加入至化合物3(15mg)的dmf(250μl)单独溶液中,并将溶液在0℃下搅拌10分钟。然后将两种溶液合并并加入额外量的hatu(2.6mg)和nmm(0.75μl),然后将反应混合物在室温下搅拌2小时。将反应溶液真空浓缩并将残余物通过反相c18柱色谱法使用洗脱剂a(v/v):水:0.05%三氟乙酸和洗脱剂b(v/v):乙腈:0.05%三氟乙酸(100:0v/v至0:100v/v)洗脱进行纯化。真空除去有机溶剂并通过冷冻干燥除去含水溶剂以得到为白色固体的化合物2(13mg)。m/z[m+2h]2+(1659,70%),[m+3h]3+(1106,100%)。
实施例2:包含氨基-6’-α-环糊精和奥瑞斯他汀环糊精有效负载mmae的缀合试剂6的合成。
使用6-单脱氧-6-单氨基-α-环糊精盐酸盐代替6-单脱氧-6-单氨基-β-环糊精盐酸盐,以与实施例1的试剂1类似的方式合成试剂6。分离为白色固体的试剂6。m/z[m+2h]2+(1578,50%),[m+3h]3+(1052,100%)。
实施例3:包含氨基-6’-γ-环糊精和奥瑞斯他汀环糊精有效负载mmae的缀合试剂7的合成。
使用6-单脱氧-6-单氨基-γ-环糊精盐酸盐代替6-单脱氧-6-单氨基-β-环糊精盐酸盐,以与实施例1的试剂1类似的方式合成试剂7。分离为白色固体的试剂7。m/z[m+2h]2+(1740,30%),[m+3h]3+(1160,100%)。
实施例4:包含氨基-3’-α-环糊精和奥瑞斯他汀环糊精有效负载mmae的缀合试剂8的合成。
使用3-单脱氧-3-单氨基-α-环糊精水合物代替6-单脱氧-6-单氨基-β-环糊精盐酸盐,以与实施例1的试剂1类似的方式合成试剂8。分离为白色固体的试剂8。m/z[m+2h]2+(1578,90%),[m+3h]3+(1052,100%)。
实施例5:包含氨基-3’-γ-环糊精和奥瑞斯他汀环糊精有效负载mmae的缀合试剂9的合成。
使用3-单脱氧-3-单氨基-γ-环糊精水合物代替6-单脱氧-6-单氨基-β-环糊精盐酸盐,以与实施例1的试剂1类似的方式合成试剂9。分离为白色固体的试剂9。m/z[m+2h]2+(1740,40%),[m+3h]3+(1160,100%)。
实施例6:包含氨基-3’-β-环糊精和奥瑞斯他汀环糊精有效负载mmae的缀合试剂10的合成。
使用3-单脱氧-3-单氨基-β-环糊精水合物代替6-单脱氧-6-单氨基-β-环糊精盐酸盐,以与实施例1的试剂1类似的方式合成试剂10。分离为白色固体的试剂10。m/z[m+2h]2+(1658,90%)。
实施例7:包含氨基-6’-β-环糊精和美登素环糊精有效负载的缀合试剂11的合成。
步骤1:化合物12的合成。
向fmoc-glu-(otbu)-oh(55mg)的dmf(1ml)溶液中加入hatu(116mg)于dmf(1ml)、nmm(34μl)中的溶液及6-单脱氧-6-单氨基-β-环糊精盐酸盐(150mg)的dmf(2ml)溶液。在室温下搅拌反应混合物16小时后,加入nmm(13μl),再过1小时后,加入另外的6-单脱氧-6-单氨基-β-环糊精盐酸盐(8mg)的dmf(200μl)溶液。3小时后,真空除去挥发物。将残余物溶解在dmf(5ml)中,并将哌啶(151μl)加入至溶液中,将溶液在室温下搅拌1小时。然后将反应溶液真空浓缩并在室温下将所得油状物沉淀到二乙基乙基(4×200ml)中,过滤得到作为白色固体的化合物12。m/z[m+h]+(1320,50%)。
步骤2:化合物13的合成。
向试剂5(156mg)的dmf(2ml)溶液中加入hatu(141mg)于dmf(1ml)、nmm(41μl)中的溶液和化合物12(196mg)的dmf(2.5ml)溶液。在0℃下搅拌2.5小时后,加入另外的试剂5(19mg)的dmf(500μl)溶液。20分钟后,将溶液真空浓缩且将残余物通过反相c18柱色谱法使用洗脱剂a(v/v):水:0.05%三氟乙酸和洗脱剂b(v/v):乙腈:0.05%三氟乙酸(100:0v/v至0:100v/v)洗脱进行纯化。真空除去有机溶剂并通过冷冻干燥除去含水溶剂以得到为无色油状物的化合物13(76mg)。m/z[m+h]+(2267,20%),[m+2h]2+(1134,100%)。
步骤3:化合物14的合成。
向化合物13(33mg)的thf:三氯甲烷(5ml,1:4v/v)溶液中加入对甲苯磺酸(14mg)并在室温下搅拌所得悬浮液。3.5小时后,真空除去挥发物并将残余物通过反相c18柱色谱法使用洗脱剂a(v/v):水:0.05%乙腈:0.05%三氟乙酸和洗脱剂b(v/v):乙腈:0.05%三氟乙酸(100:0v/v至0:100v/v)洗脱进行纯化。真空除去有机溶剂并通过冷冻干燥除去含水溶剂以得到为无色油状物的化合物14(26mg)。m/z[m+h]+(2212,25%),[m+2h]2+(1106,100%)。
步骤4:试剂11的合成。
在室温下,在惰性氩气氛下,向化合物14(18mg)的dmf(250μl)的搅拌溶液中加入hatu(6mg)。1小时后,加入另外的hatu(6mg)和nmm(1.1μl)并将溶液再搅拌0.5小时。制备val-ala-pab-ahx-dm1.tfa盐(levenabiopharma,7.5mg)和nmm(1.7μl)的dmf(100μl)的单独溶液,并在室温下搅拌20分钟,然后将两种溶液合并。加入另外的hatu(6mg)和nmm(1.7μl)并在室温下搅拌溶液。2小时后,加入另外的hatu(6mg)并将溶液在室温下再搅拌4小时,然后加入另外的nmm(1.7μl)。3.5小时后,真空除去挥发物,所得残余物通过反相c18柱色谱法使用洗脱剂a(v/v):水:5%乙腈:0.05%三氟乙酸和洗脱剂b(v/v):乙腈:0.05%三氟乙酸(100:0v/vto0:100v/v)洗脱进行纯化。真空除去有机溶剂并通过冷冻干燥除去含水溶剂以得到试剂11(2.7mg)。m/z[m+na+2h]3+(1100,65%)。[m+3h]3+(1094,100%)。
实施例8:包含氨基-6’-β-环糊精和多卡米星环糊精有效负载的缀合试剂15的合成。
步骤1:化合物16的合成。
在0℃下,向boc-val-cit-pab-多卡米星(abzena,tcrs,17mg)于无水二氯甲烷(2ml)的悬浮液中加入三氟乙酸(1ml)并在0℃下将所得溶液搅拌75分钟。然后真空除去挥发物以得到作为黄色固体的化合物16(假定定量收率,17.7mg)。m/z[m+h]+(798,100%)。
步骤2:化合物17的合成。
向化合物16(17.7mg,来自先前步骤的假定定量收率)的dmf(600μl)搅拌溶液中加入fmoc-glu(oh)-otbu(9mg)的dmf(200μl)溶液。将hatu(22mg)加入至冷却至0℃的反应混合物中,然后加入nmm(6.4μl)。20分钟后,将反应混合物加温至室温并搅拌50分钟,之后添加另外的hatu(22mg)和nmm(6.4μl)。30分钟后,将反应混合物通过反相c18柱色谱法使用洗脱剂a(v/v):水:0.05%三氟乙酸和洗脱剂b(v/v):乙腈:0.05%三氟乙酸(100:0v/v至0:100v/v)洗脱进行纯化。通过冷冻干燥除去溶剂以得到作为黄色固体的化合物17(假定定量收率,23.4mg)。m/z[m+h]+(1206,5%)。
步骤3:化合物18的合成
将化合物17(来自先前步骤的假定定量收率)溶于二氯甲烷:三氟乙酸(2.5ml2:1v/v)的溶液中。将该溶液在4℃下放置5.5小时,然后在-20℃下存储17小时。真空浓缩该溶液并将残余物通过反相c18柱色谱法使用洗脱剂a(v/v):水:0.05%三氟乙酸和洗脱剂b(v/v):乙腈:0.05%三氟乙酸(100:0v/v至0:100v/v)洗脱进行纯化。通过冷冻干燥除去溶剂以得到作为黄色固体的化合物18(3mg)。m/z[m+h]+(1149,100%)。
步骤4:化合物19的合成。
向化合物18(3mg)的dmf(220μl)溶液中加入6-单脱氧-6-单氨基-β-环糊精盐酸盐(3.7mg)和hatu(3mg)。将搅拌溶液冷却至0℃,然后加入nmm(0.9μl)。30分钟后,将溶液加温至室温,然后加入哌啶(2.6μl)并在室温下将反应混合物搅拌3.5小时。然后将反应混合物通过反相c18柱色谱法使用洗脱剂a(v/v):水:0.05%三氟乙酸和洗脱剂b(v/v):乙腈:0.05%三氟乙酸(100:0v/v至0:100v/v)洗脱进行纯化。通过冷冻干燥除去溶剂以得到作为黄色固体的化合物19(6.3mg)。m/z[m+2h]2+(1022,100%)。
步骤5:试剂15的合成
向化合物19的dmf(400μl)溶液中加入试剂5(3mg)和hatu(3.3mg)。将搅拌的溶液冷却至0℃,然后加入nmm(1μl)。1小时后,向反应混合物中加入另外的hatu(0.6mg)和nmm(0.2μl),然后将反应混合物再搅拌30分钟。然后将反应混合物通过反相c18柱色谱法使用洗脱剂a(v/v):水:0.05%三氟乙酸和洗脱剂b(v/v):乙腈:0.05%三氟乙酸(100:0v/v至0:100v/v)洗脱进行纯化。通过冷冻干燥除去溶剂以得到作为黄色固体的试剂15(3.7mg)。m/z[m+2h]2+(1495,100%),[m+na+2h]3+(1005,30%)。
实施例9:用于将试剂缀合至抗体以产生dar4的抗体药物缀合物(adc)的一般方案
将在20mm磷酸钠、ph7.5(含有150mmnacl和20mmedta)中浓度为5.2mg/ml的抗体在加热模块中加热至40℃并保持15分钟。将tcep(6当量每mab)添加至mab溶液中,轻轻混合并在40℃下温育1小时,随后使其冷却至22℃。将缀合试剂1、6、7、8、9、10、11和15溶解于dmf中以获得1.5mm溶液。用20mm磷酸钠、ph7.5(含有150mmnacl和20mmedta)将还原的mab溶液稀释至4.4mg/ml。将缀合试剂(5.6当量每mab)添加至mab溶液中,轻轻混合反应物并在22℃下温育6至22小时。然后,在22℃下,使用50mmn-乙酰基-l-半胱氨酸(20当量过量试剂)处理反应物1小时。通过疏水相互作用色谱法分析粗制缀合混合物。将粗制反应物与等体积的50mm磷酸钠、ph7(4mnacl)混合并将所得溶液上样至用50mm磷酸钠、ph7(2mnacl)平衡的toyopearlphenyl-650shic柱上。使用50mm磷酸钠、ph7(20%异丙醇)的梯度从柱上洗脱adc。合并含有dar4的adc的级分并浓缩(vivaspin20,10kdapes膜)。将浓缩的样品缓冲液更换为pbs、ph7.1-7.5并无菌过滤(0.22μmpvdf膜)。dar分配基于a248/a280吸收率。缀合物的平均dar由在280nm处的hic分析后的各个dar种类的相对峰面积计算。brentuximab抗体与缀合试剂1、6、7、8、9、10和15的缀合分别产生dar4缀合物20、21、22、23、24、25和26。抗psma抗体与试剂11的缀合产生dar4缀合物27。
实施例10:通过体外细胞存活率试验分析抗体药物缀合物(adc)通过测定对靶标过表达的癌细胞系的细胞生长的抑制作用测定实施例9制备的抗体药物缀合物20、21、22、23、24、25、26和27的体外效力。
在体外用adc或游离有效负载进行处理后的肿瘤细胞存活率的损失可通过以下方式进行测量:在增加浓度的化合物存在下培养细胞系并使用celltiter
试验中细胞系的特征和接种密度记载于下表中。我们感谢karpas博士提供了karpas-299细胞系。使用一次性neubauer计数室计数细胞,并入下表详述调整细胞密度。将karpas-299和lncap细胞以50μl/孔种植到用组织培养物处理过的不透明壁的96孔白色板中,并在37℃和5%co2下温育24小时。
在相关培养基中制备化合物的八点系列稀释液。调节每个化合物/细胞系组合的滴定范围。通过简单添加50μl/孔的2xadc稀释液处理karpas-299细胞。对于lncap细胞,除去生长培养基并用100μl/孔的1xadc稀释液代替。然后将细胞在37℃和5%co2下再培养96小时。
使用cell-titer
使用moleculardevicesspectramaxm3平板读数器记录发光,随后使用graphpadprism四参数非线性回归模型分析数据。存活率表示为未处理细胞的%并使用以下公式计算:
以%存活率相对于药物浓度的对数(以nm为单位)制图,以推断所有缀合物的ic50值。
体外细胞毒性研究的结果示于表1中。这些数据显示环糊精adc在体外具有有效的体外细胞杀伤性质。
表1
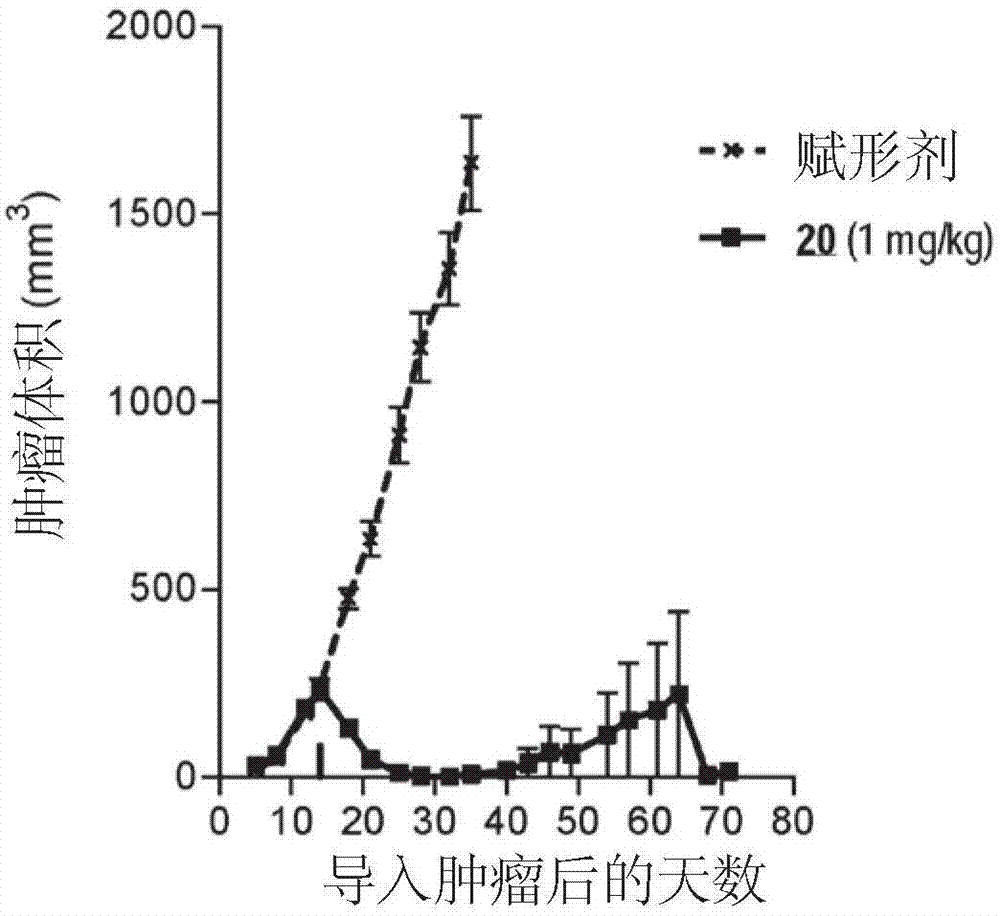
实施例11:将brentuximab-药物缀合物20、23和24与
使用平均体重为17.5g的健康雌性cb17-scid小鼠(cbysmn.cb17-prkdcscid/j,charlesriver实验室)来进行细胞接种(第0天)。在肿瘤细胞注射之前24至72小时,将小鼠进行γ-射线照射(1.44gy,60co)。动物在饲养室中在受控的环境条件下维持在依据felasa指南的spf健康状态下。
通过在右腹部皮下注射于200μlrpmi1640中的107karpas-299细胞(t-间变性大细胞淋巴瘤,alcl)来诱发肿瘤。每周两次使用卡尺测量肿瘤,使用下式估计体积:
肿瘤植入后14天(第14天),使用vivo
通过隔周的体重测量和每日对治疗相关副作用的临床体征的观察来评估治疗耐受性。当达到人道的终点时(例如1,600mm3肿瘤体积)或在给药后最多6周之后对动物实施安乐死。
对于每组,1mg/kg剂量的平均肿瘤体积±标准误差示于图1至4中。所有化合物均耐受良好。在该剂量下,缀合物20显示出总体平均肿瘤体积大大减小,且在研究期间内存活的6/8只动物中观察到完全反应(即肿瘤体积总体减少至零)。缀合物23显示出平均肿瘤体积完全消退,所有(8/8)动物在研究期间存活。缀合物24还显示出平均肿瘤体积大幅减小,在6/8只动物中具有完全反应。相比之下,市售可得的
实施例12:通过冻融试验确定抗体药物缀合物(adc)的稳定性。
通过用dpbsph7.1-7.5稀释来分别制备0.5mg/ml的adc样本20、21、22、23、24和25。
将adc样本在-80℃下温育1小时,然后在4℃下解冻。然后使用tosohbiosciencetskgelsupersw3000柱通过尺寸排阻色谱法(sec)分析样本。在用0.2m磷酸钾洗脱液、ph6.8(0.2m氯化钾和15%异丙醇)等度洗脱期间监测280nm处的uv吸光度。
表2示出了冻融试验后adc的聚集程度。源于sec分析的曲线下面积百分比(abs280)用于确定每个样本中存在的聚集物质的量。
表2
表2中的这些数据表明,每个缀合物在样本冷冻和解冻后都显示出良好的稳定性。
- 还没有人留言评论。精彩留言会获得点赞!