抑制PI3K的新型喹唑啉酮衍生物及含其的药物组合物的制作方法
本发明涉及抑制pi3k的新型喹唑啉酮衍生物以及制备这些衍生物的方法。此外,本发明提供了用于治疗血液系统恶性肿瘤、肝病或自身免疫疾病的药物组合物,所述药物组合物包含该喹唑啉酮衍生物。
背景技术:
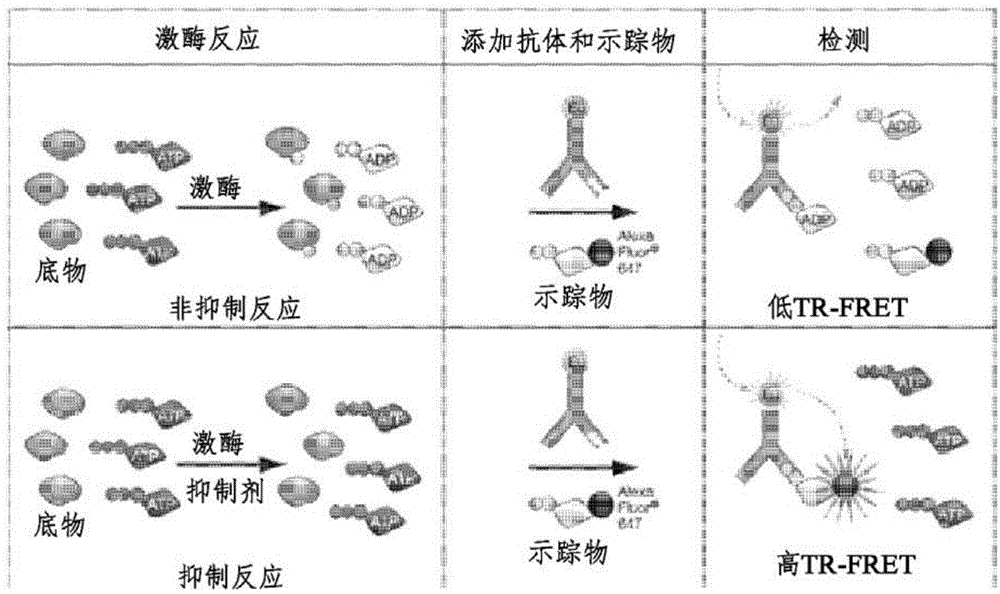
:在美国,癌症是继心脏病之后的第二大致死原因(cancerfactsandfigures2005,americancancersociety,inc.)。在癌症发展的早期阶段,可以选择通过化疗、放射疗法等去除肿瘤或杀死癌细胞的方法,但是在晚期癌症患者的情况下,由于积极治疗而引起的副作用相对较大并且治疗后的响应率低,并因此可以选择通过延缓癌症进展和改善生活质量来减少副作用的疗法。在这些方面,抗癌药物不仅旨在通过消灭癌细胞来预防癌症复发,而且当难以预期完全恢复时,还旨在通过抑制癌细胞的生长和增殖来延长生存期。现有的用于转移性癌症的化疗由于其降低的疗效而不能提供长期治疗。此外,尽管已经在医学领域引入了新的化学疗法,但仍需要新的有效药物在单一治疗或与现有药剂的共同治疗中作为一级、二级和三级疗法,用于抗性肿瘤的治疗。此外,即使具有强效力的抗癌药物也不适用于所有癌症,并因此迫切需要开发药物以提高治疗效率。靶向治疗是有利的,因为它们是肿瘤特异性的、有效的,并且相较于现有的全身性抗癌治疗,它对正常细胞的影响小得多。通常在癌细胞中发现蛋白激酶的失调,并因此它对于开发抗癌药物而言是有吸引力的靶标。在脂质激酶中,pi3k(磷脂酰肌醇-3-激酶异构体)的结构和功能近年来已经逐渐且明确地得到证实。已知pi3k属于在细胞内信号传导通路中起重要作用的酶家族,并且涉及主要的细胞功能,例如细胞生长、增殖、分化、运动、存活和胞内运输。在过去的20年中,已经稳定地揭示了当pi3k失去其调节功能时,在细胞内信号传导通路中发生诸如过度活化等问题,以导致多种类型的疾病。pi3k分为i类、ii类和iii类。i类再被划分为子类:ia类和ib类。i类pi3k处于二聚体形式,并且该二聚体分为催化亚基和调节亚基。ia类pi3k是由p110催化亚基和p85调节亚基组成的二聚体,并且就这一点而言,p110催化亚基包括三种同种型,即p110α、p110β和p110δ。因此,pi3k的同种型被称为pi3kα、pi3kβ和pi3kδ。同时,ib类pi3k是由p110γ催化亚基和p101调节亚基组成的二聚体,并且pi3k通常称为pi3kγ。pi3kδ主要由受体酪氨酸激酶(rtk)诱导而将pip2磷酸化为pip3,并且pi3kγ主要由g蛋白偶联受体(gpcr)诱导而将pip2磷酸化为pip3。pip3激活了蛋白激酶b(akt/pkb)并持续引导下游信号传导,从而参与主要的细胞功能调节,例如细胞生长、增殖、分化、运动、存活和胞内运输。近年来最为强烈的关注点之一是当pi3kδ和pi3kγ在调节胞内信号转导方面出现故障时,发生从炎症和自身免疫到血液系统恶性肿瘤和实体癌等各种疾病,并因此已经进行了大量的努力来开发通过抑制失去其调节功能的pi3kδ和pi3kγ而用于治疗炎症、自身免疫、血液系统恶性肿瘤和实体癌的药物。在该领域所开发的代表性药物的实例是艾代拉里斯(idelalisib),由gileadcalistoga开发并对pi3kδ进行选择性抑制的物质。该药物对各种类型的血液系统恶性肿瘤具有优异的疗效,并因此作为突破性药物引起了人们的注意,该药物解决了现有细胞毒性抗癌药物的问题(特别是对正常细胞的细胞毒性),并且还补偿了现有的抗癌药物的疗效问题。然而,在欧洲,临床试验期间发生了严重毒性的一些病例,其中患者死于肺炎,并因此该药物的开发目前已暂停。根据报告,原因是该药物的抑制活性对pi3kδ、而非pi3kα和pi3kβ具有足够的选择性和有效性,而且对pi3kγ缺乏足够的选择性。duvelisib对pi3kδ和pi3kγ表现出双重抑制活性,并因此有可能被开发为用于治疗血液系统恶性肿瘤、炎症和自身免疫疾病的非常有希望的药物。然而,在临床试验期间,由于类似于艾代拉里斯的问题,duvelisib的开发被终止。已知该物质的抑制效力对pi3kδ和pi3kγ的选择性不如pi3kβ。因此,需要开发能够更加选择性地对pi3kδ进行抑制,而至少不同于艾代拉里斯的药物。同时,喹唑啉酮衍生物是许多生物活性化合物中存在的特殊结构,例如作为镇静催眠药的甲喹酮、作为镇咳药的氯喹酮和作为抗惊厥药的吡甲苯喹酮(piriqualone)。喹唑啉酮及其衍生物具有广泛的生物学性质,例如催眠、止痛、抑制痉挛、抑制咳嗽和抗炎活性。特别地,喹唑啉酮衍生物被用于治疗包括癌症在内的细胞增殖性疾病,并且是近来广泛使用的治疗剂之一。例如,美国专利注册号5,747,498和5,773,476公开了喹唑啉酮衍生物,用于治疗由受体酪氨酸激酶的过度活化或异常活化导致的癌症。因此,需要通过各种方法对喹唑啉酮衍生物进行研究和开发,以用于对细胞增殖性疾病进行治疗。技术实现要素:技术问题本发明的一个目的是提供抑制pi3k的新型喹唑啉酮衍生物和制备这些衍生物的方法。本发明的另一个目的是提供包含所述喹唑啉酮衍生物的药物组合物,用于预防或治疗血液系统恶性肿瘤、肝病或自身免疫疾病。技术方案根据本申请的一个方面,提供下述式1表示的化合物或其药学上可接受的盐:[式1]在上述式1中,x是-h、卤素、-ch3或-nh2;以及y是c1-2直链烷基或c3-4环烷基。根据本发明的另一方面,提供了用于预防或治疗血液系统恶性肿瘤、肝病或自身免疫性疾病的药物组合物,所述药物组合物包含下述式1表示的化合物或其药学上可接受的盐作为活性成分:[式1]在上述式1中,x是-h、卤素、-ch3或-nh2;以及y是c1-2直链烷基或c3-4环烷基。所述血液系统恶性肿瘤可以是白血病或淋巴瘤。所述肝病可选自于由非酒精性脂肪性肝病(nafld)、非酒精性脂肪性肝炎(nash)、肝性脂肪变性、肝硬化、肝炎、肝腺瘤、胰岛素过敏症和肝癌所组成的组。所述自身免疫性疾病可选自于由过敏性鼻炎、哮喘、慢性阻塞性肺病(copd)和类风湿性关节炎所组成的组。根据本发明的又一方面,提供制备下述式1表示的化合物的方法,所述方法包括:使下述式2表示的化合物与下述式3表示的化合物反应,以制备下述式4表示的化合物;将下述式4表示的化合物脱保护,以制备下述式5表示的化合物;以及使下述式5表示的化合物与式6表示的化合物反应,以制备式1的化合物;[式1]其中,在式1中,x是-h、卤素、-ch3或-nh2;以及y是c1-2直链烷基或c3-4环烷基;[式2]其中,在式2中,x是-h、卤素、-ch3或-nh2;[式3]其中,在式3中,y是c1-2直链烷基或c3-4环烷基;[式4]其中,在式4中,x是-h、卤素、-ch3或-nh2;以及y是c1-2直链烷基或c3-4环烷基;[式5]其中,在式5中,x是-h、卤素、-ch3或-nh2;以及y是c1-2直链烷基或c3-4环烷基;[式6]其中,在式6中,z是卤素。有益效果根据本发明的新型喹唑啉酮衍生物可有效治疗血液系统恶性肿瘤或肝病。特别地,相较于现有的pi3kδ抑制剂,本发明的喹唑啉酮衍生物可以高选择性地对pi3kδ进行抑制,以显著降低免疫毒性,或同时对pi3kδ和pi3kγ进行抑制,由此不仅能够对血液系统恶性肿瘤等进行抗癌治疗,而且还能够对自身免疫性疾病进行治疗。这些靶向治疗剂可以解决诸如具有严重细胞毒性的现有抗癌治疗的副作用等问题。附图说明图1示出了实验实施例1的adapta激酶分析实验的原理。图2示出了实验实施例2的结果,用于证实对降低白血病和淋巴瘤细胞系中akt(ser473)磷酸化的作用。图3示出了实验实施例3的结果,用于证实对白血病和淋巴瘤细胞生长的抑制作用。图4示出了实验实施例4的sds-page分析结果,用于证实对弥漫性大b细胞淋巴瘤(dlbcl)细胞和急性淋巴细胞白血病(all)细胞凋亡的作用。图5示出了实验实施例5的流式细胞术结果,用于证实对弥漫性大b细胞淋巴瘤(dlbcl)细胞和急性淋巴细胞白血病(all)细胞凋亡的作用。图6示出了cytationtm5荧光显微镜(biotek)图像,其显示出实验实施例6的管形成结果,用于证实对血管生成的抑制作用。图7是显示实验实施例6中对血管生成的抑制作用的图,用于证实对血管生成的抑制作用。图8是显示实验实施例6中,用化合物处理的存活的huvec的数目的图,用于证实对血管生成的抑制作用。图9是根据实验实施例7中的式7的化合物和式8的化合物的浓度确认毒性的表,其是大鼠的单剂量毒性试验。具体实施方式在下文中,将对本发明进行详细描述。本说明书和权利要求书中使用的术语或词语不应被解释为限于普通含义或词典含义,而应当基于发明人可以适当对术语的概念进行定义而以最佳方式对发明人的发明进行解释的原则,解释为与本发明的精神一致的含义和概念。因此,在本文阐述的实施方式中描述的配置仅仅是本发明的示例性实施方式,并不代表本发明的所有技术构思,并因此应该理解的是,在本申请提交时,可以进行各种等同方式和改进方式来替换这些实施方式。本发明涉及作为pi3k抑制剂的喹唑啉酮衍生物的新型化合物。pi3k抑制剂通过与p110δ的atp结合位点对接来阻断pi3k-akt信号传导通路,并且pi3k通路的激活由pi3k催化同种型(即p110α、p110β、p110δ和p110γ)来介导。p110δ在血液系统恶性肿瘤和b细胞发育中发挥重要作用,并主要在造血干细胞中表达,并且还在多种癌症(包括白血病、淋巴瘤、结肠直肠癌、膀胱癌、恶性神经胶质瘤等)中表达。它通过pi3k-akt信号传导通路刺激相关细胞因子和趋化因子对细胞增殖进行调节。此外,根据本发明的新型喹唑啉酮衍生物克服了包括肝毒性在内的现有毒性问题。由于pi3kp110δ即使在晚期肝细胞癌中也可能高度表达,因此,根据本发明的新型喹唑啉酮衍生物作为用于肝细胞癌(实体癌)的治疗剂也是有效的。鉴于现有基于喹唑啉酮的抗癌药物的问题,新型喹唑啉酮衍生物应充分且选择性地对pi3kδ进行抑制。优选地,pi3k异构体之间的选择性需要满足以下标准:使用ic50值,pi3kα/pi3kδ和pi3kβ/pi3kδ的比例各自超过150,并且pi3kγ/pi3kδ的比例至少大于艾代拉里斯的该比例。另外,当pi3kδ和pi3kγ同时被抑制时,优选的是pi3kβ/pi3kδ和pi3kβ/pi3kγ的比例大于duvelisib的该比例。本发明提供式1表示的化合物或其药学上可接受的盐。[式1]在上述式1中,x是-h、卤素、-ch3或-nh2;以及y是c1-2直链烷基或c3-4环烷基。本文所使用的术语“卤素”是指氟(f)、溴(br)、氯(cl)或碘(i)。在式1中,y可以以(s)-异构体或(r)-异构体的形式连接,但优选以(s)-异构体的形式连接。式1化合物的具体实例包括下述式7至式14表示的化合物,但本发明不限于此。根据本发明的式7的化合物是其中x为-f且y为环丙基的式1的化合物。[式7]此外,根据本发明的式8的化合物是其中x为甲基且y为环丙基的式1的化合物。[式8]此外,根据本发明的式9的化合物是其中x为-nh2且y为环丙基的式1的化合物。[式9]此外,根据本发明的式10的化合物是其中x为-nh2且y为甲基的式1的化合物。[式10]此外,根据本发明的式11的化合物是其中x为-nh2且y为乙基的式1的化合物。[式11]此外,根据本发明的式12化合物是其中x为-cl且y为环丙基的式1的化合物。[式12]此外,根据本发明的式13的化合物是其中x为-f且y为环丁基的式1的化合物。[式13]此外,根据本发明的式14的化合物是其中x为-cl且y为环丁基的式1的化合物。[式14]由本发明的式1表示的化合物可以以药学上可接受的盐的形式使用,并且该盐可以是由药学上可接受的游离酸形成的酸加成盐。酸加成盐由以下酸获得:无机酸,如盐酸、硝酸、磷酸、硫酸、氢溴酸、氢碘酸、亚硝酸、亚磷酸等;无毒有机酸,如脂肪族单羧酸和脂肪族二羧酸、苯基取代的链烷酸、羟基链烷酸和羟基烷二酸、芳香族酸、脂肪族磺酸和芳香族磺酸等;或有机酸,如乙酸、苯甲酸、柠檬酸、乳酸、马来酸、葡萄糖酸、甲磺酸、4-甲苯磺酸、酒石酸、富马酸等。这些药学上无毒的盐的实例包括硫酸盐、焦硫酸盐、硫酸氢盐、亚硫酸盐、亚硫酸氢盐、硝酸盐、磷酸盐、磷酸一氢盐、磷酸二氢盐、偏磷酸盐、焦磷酸盐、氯化物、溴化物、碘化物、氟化物、乙酸盐、丙酸盐、癸酸盐、辛酸盐、丙烯酸盐、甲酸盐、异丁酸盐、癸酸盐、庚酸盐、丙炔酸盐、草酸盐、丙二酸盐、琥珀酸盐、辛二酸盐、癸二酸盐、富马酸盐、马来酸盐、丁炔-1,4-二酸盐、己烷-1,6-二酸盐、苯甲酸盐、氯苯甲酸盐、甲基苯甲酸盐、二硝基苯甲酸盐、羟基苯甲酸盐、甲氧基苯甲酸盐、邻苯二甲酸盐、对苯二甲酸盐、苯磺酸盐、甲苯磺酸盐、氯苯磺酸盐、二甲苯磺酸盐、苯基乙酸盐、苯基丙酸盐、苯基丁酸盐、柠檬酸盐、乳酸盐、β-羟基丁酸盐、乙醇酸盐、苹果酸盐、酒石酸盐、甲磺酸盐、丙磺酸盐、萘-1-磺酸盐、萘-2-磺酸盐、扁桃酸盐等。可以使用常规方法来制备根据本发明的酸加成盐。例如,这些酸加成盐可以通过以下来制备:将式1的衍生物溶解在有机溶剂(例如甲醇、乙醇、丙酮、二氯甲烷、乙腈等)中,向其中加入有机酸或无机酸,以生成沉淀物,以及过滤并干燥沉淀物;或者可以通过以下来制备:在减压下对溶剂和过量的酸进行蒸馏,然后干燥得到的溶液,随后在有机溶剂的存在下进行结晶。此外,可以通过使用碱来制备药学上可接受的金属盐。碱金属或碱土金属盐通过以下获得,例如将化合物溶解在过量的碱金属氢氧化物或碱土金属氢氧化物溶液中,过滤不溶性化合物盐,以及蒸发并干燥滤液。此时,药学上优选制备钠盐、钾盐或钙盐作为金属盐。另外,通过使碱金属或碱土金属盐与合适的银盐(例如硝酸银)反应,得到与其相应的盐。此外,本发明不仅包括式1表示的化合物及其药学上可接受的盐,还包括可由其制备的溶剂化物、立体异构体和水合物等。本发明还提供用于预防或治疗血液系统恶性肿瘤、肝病和自身免疫疾病的药物组合物,所述药物组合物包含下述式1表示的化合物或其药学上可接受的盐作为活性成分。[式1]在上述式1中,x是-h、卤素、-ch3或-nh2;以及y是c1-2直链烷基或c3-4环烷基。在式1中,y可以以(s)-异构体或(r)-异构体的形式连接,但优选y以(s)-异构体的形式连接。在根据本发明的药物组合物中,式1表示的化合物或其药学上可接受的盐可以在临床给药期间以各种剂型口服给药或肠胃外给药,并且可以使用常用的稀释剂或赋形剂来配制,如填充剂、增量剂、粘合剂、润湿剂、崩解剂和表面活性剂等。用于口服给药的制剂可包括例如片剂、丸剂、硬胶囊/软胶囊、液体剂、混悬剂、乳剂、糖浆剂、颗粒剂、酏剂、混悬剂、锭剂等。除活性成分外,这些制剂还包含稀释剂(例如乳糖、右旋糖、蔗糖、甘露醇、山梨糖醇、纤维素和/或甘氨酸)或润滑剂(例如二氧化硅、滑石粉、硬脂酸及其镁盐或钙盐、和/或聚乙二醇)。片剂可包含粘合剂,例如硅酸铝镁、淀粉糊、明胶、甲基纤维素、羧甲基纤维素钠和/或聚乙烯吡咯烷酮,并且在一些情况下,片剂可以包含崩解剂(例如淀粉、琼脂、海藻酸或其钠盐)、或沸腾混合物(boilingmixture)和/或吸附剂、着色剂、矫味剂和甜味剂。包含式1表示的化合物作为活性成分的药物组合物可以肠胃外给药,并且通过皮下注射、静脉内注射、肌内注射或胸腔内注射进行肠胃外给药。在这方面,为了制备用于肠胃外给药的制剂,将式1表示的化合物或其药学上可接受的盐与稳定剂或缓冲剂在水中混合,以制备溶液或混悬剂,然后制备成安瓿或小瓶单位剂型。所述组合物可以是灭菌的和/或可以包含辅助剂(例如防腐剂、稳定剂、可湿性粉剂、渗透调节用盐和/或缓冲剂)、以及其它治疗上有效的物质,并且可以使用常规方法(例如混合、制粒或包衣)进行配制。除喹唑啉酮化合物外,本发明的组合物还可包含表现出相同或相似功能的一种或多种有效成分。可以根据患者的病情和体重、症状的严重程度、剂型、给药途径和给药时间适当选择合适剂量的本发明的药物组合物。在本发明的组合物中,为了最佳功效,优选每天以0.2mg/kg至200mg/kg的量给予有效成分。所述组合物可以每天给药一次或每天给药多次,但本发明不限于此。根据本发明,所述血液系统恶性肿瘤可以是白血病或淋巴瘤。所述白血病可选自急性淋巴细胞白血病(all)、急性髓性白血病(aml)、慢性淋巴细胞白血病(cll)和小淋巴细胞淋巴瘤(sll),并且急性淋巴细胞白血病也称为急性成淋巴细胞性白血病。淋巴瘤可以是成熟(外周)b细胞肿瘤,并且更具体地,可以选自b细胞慢性淋巴细胞白血病/小淋巴细胞淋巴瘤;b细胞幼淋巴细胞白血病;淋巴浆细胞性淋巴瘤;边缘区淋巴瘤,例如,脾边缘区b细胞淋巴瘤(+/-绒毛淋巴细胞)、淋巴结边缘区淋巴瘤(+/-单核细胞样b细胞)和粘膜相关淋巴组织(malt)型淋巴结外边缘区b-细胞淋巴瘤;毛细胞白血病;浆细胞骨髓瘤/浆细胞瘤;滤泡性淋巴瘤;卵泡中心淋巴瘤;套细胞淋巴瘤;弥漫性大b细胞淋巴瘤(包括纵隔大b细胞淋巴瘤、血管内大b细胞淋巴瘤和原发性渗出性淋巴瘤);以及伯基特(burkitt)淋巴瘤/伯基特细胞淋巴瘤。此外,淋巴瘤可选自多发性骨髓瘤(mm)、非霍奇金淋巴瘤(nhl)、套细胞淋巴瘤(mcl)、滤泡性淋巴瘤、瓦尔登斯特伦(waldenstrom)巨球蛋白血症(wm)、b细胞淋巴瘤和弥漫性大b-细胞淋巴瘤(dlbcl)。本发明的肝病可选自于由非酒精性脂肪性肝病(nafld)、非酒精性脂肪性肝炎(nash)、肝性脂肪变性、肝硬化、肝炎、肝腺瘤、胰岛素过敏症和肝癌所组成的组。所述肝癌可以是例如肝肿瘤、肝细胞腺瘤或肝细胞癌。自身免疫疾病可选自于由过敏性鼻炎、哮喘、慢性阻塞性肺病(copd)和类风湿性关节炎所组成的组。本发明还提供了制备下述式1表示的化合物的方法,其中,所述方法包括:使下述式2表示的化合物与下述式3表示的化合物反应,以制备下述式4表示的化合物;将式4的化合物脱保护,以制备下述式5表示的化合物;以及使下述式5表示的化合物与式6表示的化合物反应,以制备式1表示的化合物;[式1]其中,在式1中,x是-h、卤素、-ch3或-nh2;以及y是c1-2直链烷基或c3-4环烷基;[式2]其中,在式2中,x是-h、卤素、-ch3或-nh2;[式3]其中,在式3中,y是c1-2直链烷基或c3-4环烷基;[式4]其中,在式4中,x是-h、卤素、-ch3或-nh2;以及y是c1-2直链烷基或c3-4环烷基;[式5]其中,在式5中,x是-h、卤素、-ch3或-nh2;以及y是c1-2直链烷基或c3-4环烷基;[式6]其中,在式6中,z是卤素。步骤1是通过使式2表示的化合物与式3表示的化合物反应制备式4表示的化合物的过程。例如,在室温下搅拌的同时,可以将亚磷酸三苯酯加入至在吡啶溶剂的存在下的式2表示的化合物和式3表示的化合物混合在一起的溶液中。此时,温度无特别限制,但混合物可以在30℃至100℃、优选45℃至80℃、并且更优选55℃至60℃的温度下进行搅拌。搅拌时间无特别限制,但搅拌过程可以进行5小时至20小时、优选8小时至16小时、并且更优选10小时至14小时。随后,可以加入苯胺以使反应发生。此时,温度无特别限制,但反应可以在50℃至200℃、优选90℃至150℃、并且更优选100℃至120℃的温度下进行。此时,反应时间无特别限制,但反应可以进行1小时至20小时、优选3小时至15小时、并且更优选5小时至10小时。步骤2是通过将式4表示的化合物脱保护来制备式5表示的化合物的过程。例如,式5表示的化合物可以制备如下:将式4表示的化合物加入至其中溶解有三氟乙酸(cf3cooh)的二氯甲烷溶液中,然后在室温下反应0.1小时至2小时、优选0.2小时至1.5小时、并且更优选0.5小时至1小时。步骤3是通过使式5表示的化合物与式6表示的化合物反应制备式1表示的化合物的过程。例如,将式5表示的化合物加入到叔丁醇中,向其中加入n,n-二异丙基乙胺,然后将式6表示的化合物加入到所得的溶液中,并且可以在回流的同时将反应溶液搅拌10小时至48小时、优选15小时至30小时、并且更优选20小时至26小时。本发明提供了用于预防或缓解血液系统恶性肿瘤或肝病的保健功能食品,所述保健功能食品包含新型喹唑啉酮化合物或其药学上可接受的盐作为活性成分。所述保健功能食品可以制备成饮料、口香糖、茶、糖果、维生素复合物和保健补品等各种形式,但不限于此。在下文中,将参考实施例和实验实施例对本发明进行详细描述。然而,提供这些实施例和实验实施例仅用于说明目的,并不旨在对本发明的范围进行限制。实施例1:(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮(式7)的制备[反应式1]步骤1:(s)-叔丁基环丙基(5-氟-4-氧代-3-苯基-3,4-二氢喹唑啉-2-基)甲基氨基甲酸酯的制备向其中将2-氨基-6-氟苯甲酸(1.0当量)和(s)-2-(叔丁氧基羰基氨基)-2-环丙基乙酸(1.0当量)混合在吡啶溶剂中的溶液中加入亚磷酸三苯酯(1.4当量),同时在室温下搅拌该溶液。将所得混合物在55℃至60℃下搅拌12小时。向其中加入苯胺(1.4当量),然后在110℃左右反应7小时。此后,将混合的反应溶液冷却至室温并用乙酸乙酯和水萃取。将得到的有机层用无水硫酸镁(mgso4)脱水,并在减压下浓缩。将正庚烷加入到残余物中,然后搅拌30分钟以析出固体,将固体过滤并用正庚烷洗涤,然后将得到的固体干燥,以得到(s)-叔丁基环丙基(5-氟-4-氧代-3-苯基-3,4-二氢喹唑啉-2-基)甲基氨基甲酸酯,产率为65%至80%。1hnmr(300mhz,cdcl3):δ7.66-7.73(m,1h),7.50-7.61(m,4h),7.32-7.40(m,2h),7.09-7.15(t,j=18hz,1h),5.53-5.56(d,j=9hz,1h),4.18-4.23(t,j=15hz,1h),1.42(s,9h),1.08-1.16(m,1h),0.38-0.42(m,2h),0.24-0.30(m,1h),0.01-0.11(m,1h)。步骤2:(s)-2-(氨基(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮的制备将三氟乙酸(约为(s)-叔丁基环丙基(5-氟-4-氧代-3-苯基-3,4-二氢喹唑啉-2-基)甲基氨基甲酸酯的重量的8倍)加入到溶解有(s)-叔丁基环丙基(5-氟-4-氧代-3-苯基-3,4-二氢喹唑啉-2-基)甲基氨基甲酸酯的二氯甲烷溶液(约为(s)-叔丁基环丙基(5-氟-4-氧代-3-苯基-3,4-二氢喹唑啉-2-基)甲基氨基甲酸酯的重量的15倍)。将反应溶液在室温下搅拌约0.5小时至约1小时,然后使用碳酸钠水性溶液将反应溶液的ph调节至约7。将二氯甲烷溶液分离,使用硫酸镁(mgso4)脱水并过滤,以及除去硫酸镁(mgso4),然后将滤液减压浓缩,以得到(s)-2-(氨基(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮,产率为80%至95%。1hnmr(400mhz,cdcl3):δ7.66-7.71(m,1h),7.47-7.57(m,4h),7.27-7.31(m,2h),7.08-7.12(t,j=16hz,1h),2.97-2.99(d,j=8hz,1h),1.87(s,2h),1.22-1.31(m,1h),0.39-0.53(m,2h),0.01-0.15(m,2h)。步骤3:(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮的制备将步骤2中得到的(s)-2-(氨基(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮加入到叔丁醇(约为(s)-2-(氨基(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮的重量的15倍),向其中加入n,n-二异丙胺(约为(s)-2-(氨基(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮的2当量)和6-溴-9h-嘌呤,然后在回流的同时将反应溶液搅拌24小时。将反应混合物冷却并减压浓缩以除去叔丁醇。向浓缩物中加入乙酸乙酯,并依次用稀盐酸溶液和稀碳酸钾溶液洗涤。将乙酸乙酯层用无水硫酸镁(mgso4)脱水和过滤,并将滤液减压浓缩,以得到处于固体形式的(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮(式7),产率为60%至80%。1hnmr(300mhz,cdcl3):δ13.02(s,1h),8.03(s,1h),7.98(s,1h),7.50-7.71(m,6h),7.39-7.42(dd,j=9hz,1h),7.08-7.14(t,j=18hz,1h),6.76-6.79(d,j=9hz,1h),4.93(brs,1h),1.72(brs,1h),1.33-1.44(m,1h),0.49-0.53(m,2h),0.37-0.46(m,1h),0.21-0.27(m,1h)。esi-msm/z428.45[m+h]+实施例2:(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-甲基-3-苯基喹唑啉-4(3h)-酮(式8)的制备[反应式2]步骤1:(s)-叔丁基环丙基(5-甲基-4-氧代-3-苯基-3,4-二氢喹唑啉-2-基)甲基氨基甲酸酯的制备除了使用2-氨基-6-甲基苯甲酸代替2-氨基-6-氟苯甲酸之外,以与实施例1相同的方式制备(s)-叔丁基环丙基(5-甲基-4-氧代-3-苯基-3,4-二氢喹唑啉-2-基)甲基氨基甲酸酯。1hnmr(400mhz,cdcl3):δ8.33(br.s.,3h),7.50(d,4h,j=7.9hz),7.28(t,11h,j=7.7hz),7.07(t,2h,j=7.3hz),5.35(br.s.,1h),3.61(br.s.,3h),1.32-1.51(m,12h),1.17-1.30(m,1h),0.51-0.73(m,4h),0.47(td,3h,j=4.7,9.6hz)。步骤2:(s)-2-(氨基(环丙基)甲基)-5-甲基-3-苯基喹唑啉-4(3h)-酮的制备以与实施例1相同的方式制备(s)-2-(氨基(环丙基)甲基)-5-甲基-3-苯基喹唑啉-4(3h)-酮。1hnmr(400mhz,dmso-d6):δ8.41(br.s.,2h),7.79(t,1h,j=7.7hz),7.54-7.73(m,2h),7.31-7.46(m,1h),2.74(s,3h),1.23(br.s.,1h),1.18(tt,1h,j=4.4,8.7hz),0.51(s,1h),0.32-0.41(m,1h,j=4.8,10,10hz)。步骤3:(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-甲基-3-苯基喹唑啉-4(3h)-酮的制备除了使用(s)-2-(氨基(环丙基)甲基)-5-甲基-3-苯基喹唑啉-4(3h)-酮代替(s)-2-(氨基(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮之外,以与实施例1相同的方式制备(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-甲基-3-苯基喹唑啉-4(3h)-酮(式8)。1hnmr(400mhz,cdcl3):δ8.29(s,1h),7.96(br.s.,1h),7.36-7.71(m,7h),7.19-7.25(m,1h),6.83(d,1h,j=6.6hz),4.96(t,1h,j=8.1hz),2.82(s,3h),1.24-1.43(m,2h),0.29-0.67(m,3h),0.24(s,1h),0.07(s,1h)。esi-msm/z424.48[m+h]+实施例3:(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-氨基-3-苯基喹唑啉-4(3h)-酮(式9)的制备[反应式3]步骤1:(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-((4-甲氧基苄基)氨基)-3-苯基喹唑啉-4(3h)-酮的制备向密封管中容纳通过向乙醇(三乙胺的体积的15倍)中依次加入上述实施例1制备的(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)(1.0当量)和三乙胺(5.0当量)制备的溶液,再加入4-甲氧基苄胺。随后,用氮气对管进行置换并密封,然后将反应混合物加热至180℃并反应一天。冷却至室温后,减压除去乙醇溶剂。此后,使粗混合物经受硅胶柱色谱(二氯甲烷/甲醇20:1)纯化,以得到作为黄色固体的(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-((4-甲氧基苄基)氨基)-3-苯基喹唑啉-4(3h)-酮(收率:38%)。1hnmr(300mhz,cdcl3):δ13.67(s,1h),8.80-8.84(t,j=12hz,1h),8.31(s,1h),7.96(s,1h),7.52-7.62(m,4h),7.39-7.48(m,2h),7.23-7.25(d,j=6hz,2h),6.84-6.92(t,j=24hz,2h),6.80-6.84(d,j=12hz,2h),6.46-6.49(d,j=9hz,1h),4.92(s,1h),4.31-4.33(d,j=6hz,2h),3.76(s,3h),1.37-1.39(m,1h),0.43-0.50(m,2h),0.38-0.40(m,1h),0.20-0.25(m,1h)。步骤2:(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-氨基-3-苯基喹唑啉-4(3h)-酮的制备向溶解有(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-((4-甲氧基苄基)氨基)-3-苯基喹唑啉-4(3h)-酮(1.0当量)的二氯甲烷((s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-((4-甲氧基苄基)氨基)-3-苯基喹唑啉-4(3h)-酮的体积的6倍)的溶液中加入三氟乙酸(二氯甲烷的体积的2倍),并将得到的溶液在室温下搅拌0.5小时至2小时。此后,在0℃下用1mnaoh溶液将粗混合物的ph调节至7。将所得溶液用二氯甲烷萃取三次,并将经合并的有机相用无水硫酸镁(mgso4)脱水,并在减压下浓缩。将残余物通过硅胶柱色谱进行纯化,以得到作为象牙色固体的(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-氨基-3-苯基喹唑啉-4(3h)-酮(式9)(收率:21%)。1hnmr(300mhz,cdcl3):δ13.16(s,1h),8.30(s,1h),7.97(s,1h),7.53-7.64(m,4h),7.40-7.45(t,j=15hz,2h),6.92-6.95(d,j=9hz,1h),6.84-6.86(d,j=6hz,1h),6.54-6.56(d,j=6hz,1h),6.15(s,2h),4.93(s,1h),1.32-1.41(m,1h),0.47-0.48(m,2h),0.38-0.43(m,1h),0.22-0.25(m,1h)。实施例4:(s)-2-(1-((7h-嘌呤-6-基)氨基)乙基)-5-氨基-3-苯基喹唑啉-4(3h)-酮(式10)的制备[反应式4]除了使用(s)-2-(1-((7h-嘌呤-6-基)氨基)乙基)-5-氟-3-苯基喹唑啉-4(3h)-酮代替(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮之外,以与实施例3相同的方式制备(s)-2-(1-((7h-嘌呤-6-基)氨基)乙基)-5-氨基-3-苯基喹唑啉-4(3h)-酮(式10)。1hnmr(500mhz,dmso-d6):δ8.22-8.00(m,1h),7.78-7.32(m,4h),7.06(br.s.,1h),6.70-6.55(m,1h),1.99(s,1h),1.55-1.06(m,4h),0.93-0.70(m,2h)。实施例5:(s)-2-(1-(7h-嘌呤-6-基氨基)丙基)-5-氨基-3-苯基喹唑啉-4(3h)-酮(式11)的制备[反应式5]除了使用(s)-2-(1-((7h-嘌呤-6-基)氨基)丙基)-5-氟-3-苯基喹唑啉-4(3h)-酮代替(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮之外,以与实施例3相同的方式制备(s)-2-(1-(7h-嘌呤-6-基氨基)丙基)-5-氨基-3-苯基喹唑啉-4(3h)-酮(式11)。1hnmr(300mhz,cdcl3):δ8.31(s,1h),7.97(s,1h),7.35-7.68(m,6h),6.91-6.94(d,j=9hz,1h),6.82-6.84(d,j=6hz,1h),6.53-6.56(d,j=9hz,1h),6.15(s,2h),5.16(s,1h),1.91-2.05(m,1h),1.74-1.84(m,1h),0.84-0.89(t,j=15hz,3h)。实施例6:(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-氯-3-苯基喹唑啉-4(3h)-酮(式12)的制备[反应式6]步骤1:(s)-叔丁基(5-氯-4-氧代-3-苯基-3,4-二氢喹唑啉-2-基)(环丙基)甲基氨基甲酸酯的制备除了使用2-氨基-6-氯苯甲酸代替2-氨基-6-氟苯甲酸之外,以与实施例1相同的方式制备(s)-叔丁基(5-氯-4-氧代-3-苯基-3,4-二氢喹唑啉-2-基)(环丙基)甲基氨基甲酸酯。步骤2:(s)-2-(氨基(环丙基)甲基)-5-氯-3-苯基喹唑啉-4(3h)-酮的制备以与实施例1相同的方式制备(s)-2-(氨基(环丙基)甲基)-5-氯-3-苯基喹唑啉-4(3h)-酮。步骤3:(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-氯-3-苯基喹唑啉-4(3h)-酮的制备除了使用(s)-2-(氨基(环丙基)甲基)-5-氯-3-苯基喹唑啉-4(3h)-酮代替(s)-2-(氨基(环丙基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮之外,以与实施例1相同的方式制备(s)-2-(((7h-嘌呤-6-基)氨基)(环丙基)甲基)-5-氯-3-苯基喹唑啉-4(3h)-酮(式12)。1hnmr(300mhz,cdcl3):δ13.02(s,1h),8.02(s,1h),7.98(s,1h),7.15-7.68(m,8h),6.76-6.79(d,j=9hz,1h),4.93(br.s.,1h),1.72(br.s.,1h),1.33-1.44(m,1h),0.49-0.53(m,2h),0.37-0.46(m,1h),0.21-0.27(m,1h)。esi-msm/z444.40[m+h]+实施例7:(s)-2-(((7h-嘌呤-6-基)氨基)(环丁基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮(式13)的制备除了使用(s)-2-(叔丁氧基羰基氨基)-2-环丁基乙酸代替(s)-2-(叔丁氧基羰基氨基)-2-环丙基乙酸之外,以与实施例1相同的方式制备(s)-2-(((7h-嘌呤-6-基)氨基)(环丁基)甲基)-5-氟-3-苯基喹唑啉-4(3h)-酮(式13)。1hnmr(300mhz,dmso-d6):δ12.95(s,1h),8.13(br.s.,1h),7.85(br.s.,1h),7.24-7.60(m,8h),5.18(br.s.,1h),3.05(br.s.,1h),1.64-2.01(m,7h)。实施例8:(s)-2-(((7h-嘌呤-6-基)氨基)(环丁基)甲基)-5-氯-3-苯基喹唑啉-4(3h)-酮(式14)的制备除了使用2-氨基-6-氯苯甲酸代替2-氨基-6-氟苯甲酸并使用(s)-2-(叔丁氧基羰基氨基)-2-环丁基乙酸代替(s)-2-(叔丁氧基羰基氨基)-2-环丙基乙酸之外,以与实施例1相同的方式制备(s)-2-(((7h-嘌呤-6-基)氨基)(环丁基)甲基)-5-氯-3-苯基喹唑啉-4(3h)-酮(式14)。1hnmr(300mhz,dmso-d6):δ12.88(s,1h),8.17(br.s.,1h),8.00(s,1h),7.12-7.87(m,8h),5.18(br.s.,1h),3.06(br.s.,1h),1.62-1.99(m,7h)。式7至式14的化合物的结构示于下表1中。表1实验实施例1:pi3k激酶活性试验(1)实验方法使用均相的基于荧光的免疫测定进行实验,该免疫测定是adapta激酶测定。进行可从thermofisherscientificinc.获得的selectscreentm服务。实验原理在图1中示出。(2)实验结果实验结果在下表2中示出。如表2所示,式7的化合物和式8的化合物表现出比对照药物艾代拉里斯更高的活性。特别是,它们对p110δ表现出特异性抑制活性,并且对p110γ也表现出优异的活性。特别地,在式7的化合物的情况下,pi3kα/pi3kδ和pi3kβ/pi3kδ的比例(作为ic50值)分别为412和210;在式8的化合物的情况下,pi3kα/pi3kδ和pi3kβ/pi3kδ的比例(作为ic50值)分别为1,488和1,800。此外,证实了式7的化合物和式8的化合物具有高δ(δ)选择性。它们各自分别表现出pi3kγ/pi3kδ的比例为51和95,而艾代拉里斯则表现出约25的该比例。由这些结果证实,式7的化合物和式8的化合物可对δ(δ)依赖性癌症具有有效且足够的活性。表2ic50(nm)ic50(nm)ic50(nm)ic50(nm)pi3kp110αp110βp110γp110δ艾代拉里斯498570230.9式720610525.70.5式81341628.60.09实验实施例2:证实对降低白血病和淋巴瘤细胞系中的akt(ser473)磷酸化的作用的实验(1)实验方法使细胞系(sudhl5、sudhl10、ccrf-sb和molt4)经受血清耗竭2小时,然后用1μm的式7的化合物、式8的化合物、艾代拉里斯(对照药物1)、tgr1202(对照药物2)和二甲基亚砜(dmso)各自处理1小时。随后,细胞裂解并根据大小进行分级,然后用针对磷酸化akt(ser473)的抗体进行免疫印迹。(2)实验结果实验结果在图2中示出。如图2所示,证实了式7的化合物和式8的化合物在各种弥漫性大b细胞淋巴瘤(dlbcl)和急性淋巴细胞白血病(all)细胞中诱导akt磷酸化的降低。实验实施例3:证实抑制白血病和淋巴瘤细胞生长的作用的实验(1)实验方法pi3kp110δ在白血病和淋巴瘤细胞系中高度表达,并且通过抑制pi3kp110δ来抑制细胞生长。因此,进行该实验以证实化合物对抑制细胞生长的作用。将弥漫性大b细胞淋巴瘤(dlbcl)来源的细胞和急性淋巴细胞白血病(all)来源的细胞与式7的化合物、式8的化合物或艾代拉里斯随同对照培养基一起培养48小时。通过测量细胞计数试剂盒-8(cck-8)染料的吸光度评价对dlbcl来源的细胞和all来源的细胞的细胞生长抑制作用。在48小时的最后3小时期间,向每个板中加入10μmcck-8染料,然后进行培养。所有数据以三次独立实验的平均值(±sd)表示。(2)实验结果实验结果在图3中示出。如图3所示,dlbcl来源的细胞和all来源的细胞系的生长在0.625μm至20μm的浓度范围内降低。此时,将致死浓度50(lc50)(即,使50%细胞致死的化合物浓度)示于表3中,并且证实了式7的化合物和式8的化合物表现出低于艾代拉里斯的lc50值。表3lc50(μm)lc50(μm)lc50(μm)细胞系sudhl5(dlbcl)molt4(all)supb15(all)艾代拉里斯3.0>20>20式71.113.7>20式80.94.916.53实验实施例4:通过sds-page证实对弥漫性大b细胞淋巴瘤(dlbcl)和急性淋巴细胞白血病(all)细胞凋亡的作用的实验(1)实验方法将2.6×106个细胞与浓度为50μm的式8的化合物、式7的化合物或艾代拉里斯(对照药物)培养36小时,然后通过十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sds-page)进行蛋白质分析。半胱天冬酶3和半胱天冬酶9parp蛋白是参与细胞凋亡的蛋白质,通常作为无活性前体(fl)存在,并且当收到凋亡刺激信号时,它们通过裂解(cl)而被激活。使用这些抗体进行免疫印迹分析,并使用甘油醛-3-磷酸脱氢酶(gapdh)作为上样对照。(2)实验结果实验结果在图4中示出。如图4所示,式7的化合物和式8的化合物在弥漫性大b细胞淋巴瘤(dlbcl)和急性淋巴细胞白血病(cll)细胞中诱导细胞凋亡。此外,相较于对照药物(艾代拉里斯),伴随着式7的化合物和式8的化合物的细胞凋亡更加活跃地发生。从这些结果证实,式7的化合物和式8的化合物通过细胞凋亡来抑制细胞生长。实验实施例5:通过流式细胞术证实对弥漫性大b细胞淋巴瘤(dlbcl)和急性淋巴细胞白血病(all)细胞凋亡的作用的实验(1)实验方法将1×106个弥漫性大b细胞淋巴瘤(dlbcl)细胞和1×106个急性淋巴细胞白血病(all)细胞进行培养,并用对照药物(艾代拉里斯)、式7的化合物和式8的化合物各自以50μm的浓度处理24小时。用pbs对细胞进行洗涤,然后悬浮在结合缓冲液中。向其中加入5μl膜联蛋白v-fitc储备溶液(bectondickinsonscience,inc)和5μlpi(20μg/ml),随后在室温下随着遮光而孵育15分钟,然后通过流式细胞术在facscantm(bectondickinson)上对靶标材料进行定量。(2)实验结果实验结果在图5中示出。如图5所示,证实了相较于对照药物(艾代拉里斯),式7的化合物和式8的化合物更有效地诱导了细胞凋亡。实验实施例6:证实对血管生成的抑制作用的实验(1)实验方法为了比较式7的化合物、式8的化合物和对照药物(艾代拉里斯)的血管生成抑制的水平,人脐静脉内皮细胞(huvec)(血管内皮细胞)和内皮细胞生长培养基获得自lifetechnologies。在37℃下,将人脐静脉内皮细胞(huvec)连同式7的化合物、式8的化合物或对照药物(艾代拉里斯)一起在基底膜基质上进行培养。18小时后,使用cytationtm5荧光显微镜(biotek)拍摄管形成,并使用软件计算由分支点形成的区域的数量(参见图6)。(2)实验结果实验结果在图7和图8中示出。如图7所示,相较于对照药物(艾代拉里斯),式7的化合物和式8的化合物更能抑制血管生成。另外,如图8所示,式7的化合物和式8的化合物不会显著地影响人脐静脉内皮细胞(huvec)的细胞毒性。实验实施例7:大鼠的单剂量毒性试验(1)实验方法将式7的化合物和式8的化合物经口服给予至8只6周龄雌性大鼠,以观察其单剂量口服毒性,并获得近似致死剂量。剂量设定为10ml/kg,并且基于体重计算每只大鼠的剂量。每种化合物以100mg/kg、300mg/kg、900mg/kg和1,500mg/kg的剂量给药,并且在给药后第1天至第2天每天一次观察全身症状。(2)实验结果实验结果在图9中示出。式7的化合物的致死剂量超过1,500mg/kg,并且式8的化合物的致死剂量为1,500mg/kg。同时,根据本发明的式1表示的化合物可以配制成各种形式。以下提供使用根据本发明的式1表示的化合物作为活性成分的数种配制方法仅用于说明目的,而不旨在对本发明进行限制。制备实施例1:药物制剂的配制1-1.粉剂的制备式1的化合物500mg乳糖100mg滑石粉10mg将上述成分混合,并用其填充气密包装,以制备粉剂。1-2.片剂的制备将上述成分混合,然后根据制备片剂的一般方法制备片剂。1-3.胶囊的制备将上述成分混合,然后根据制备胶囊的一般方法用其填充明胶胶囊以制备胶囊。1-4.注射剂的制备式1的化合物500mg注射用无菌蒸馏水适量ph调节剂适量根据制备注射剂的一般方法,以在单个安瓿(2ml)中包含上述成分来制备一次用量的针剂。1-5.液体剂的制备根据制备液体剂的一般方法,将各成分加入并溶解在纯净水中。加入适量的柠檬香料,并将上述成分混合。向其中加入纯化水,从而使所得溶液的总量调节至100ml。用其填充棕色瓶子,并进行灭菌以制备液体剂。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1