一种siRNA和抗癌药物疏水性复合物及其制备方法与应用与流程
本发明涉及核酸药物和化疗药物共递送领域,具体涉及一种sirna和抗癌药物疏水性复合物及其制备方法与应用。
背景技术:
选择性下调靶基因表达的rna干扰技术(rnai)正成为治疗癌症的最有前途的方法之一,rnai在体内的成功应用一般通过纳米载体介导的小干扰rna(sirna)的传递来实现。一些有力的证据表明,sirna和传统化疗药物同时递送具有协同抗癌的作用。到目前为止,阳离子脂质和基于聚合物的纳米载体仍是sirna的主要传递系统。为了实现两种药物的共递送,sirna通常通过静电作用荷载到带正电荷的纳米载体上,而化疗药物则通过物理包封或化学结合加载到纳米载体上。近几十年来尽管付出了很多努力,这些载有sirna的阳离子纳米载体的临床转化仍然面临着各种制剂方面的挑战,包括纳米载体设计复杂,难以扩大生产和潜在的毒性等问题。作为一种替代策略,用临床批准的聚(乙二醇)-嵌段-聚(d,l-丙交酯)(peg-b-pla)胶束作为递送载体可能是加速临床转化的有效方法。然而,sirna固有的物理化学特性,包括高分子量,负电性和极端亲水性,使其难以加载到非阳离子纳米载体中。因此,迫切需要探索一种有效且方便的方法通过使用peg-b-pla胶束对sirna和抗癌药物进行共递送。
技术实现要素:
为了克服现有技术的不足,本发明的一个目的是提供一种sirna和抗癌药物疏水性复合物及其制备方法。
本发明的疏水性sirna和抗癌药物疏水性复合物按照下述方法制备:将不同浓度的抗癌药物(如dox)·hcl逐渐加入到fam-sirna的水溶液中,抗癌药物与sirna核苷酸(sirna-nt)单位设置不同摩尔比,在室温下温育后,将混合物溶液离心以收集形成的[sirna和抗癌药物]复合物的hip。
本发明的另一个目的是制备荷载hip[sirna&抗癌药物]纳米颗粒。
本发明所提供的纳米颗粒,包载了用hip法制备的[sirna和抗癌药物]疏水复合物。并从细胞水平和动物水平验证了np[sirna和dox]协同抗肿瘤效果。
本发明的目的通过以下技术方案实现。
一种sirna和抗癌药物疏水性复合物的制备方法,包括以下步骤:
将水溶性抗癌药加入到sirna水溶液中,室温孵育,离心收集形成了sirna和抗癌药物疏水性复合物,标记为hip[sirna&抗癌药物]复合物。
优选的,步骤(1)所述水溶性抗癌药为盐酸多柔比星、盐酸表柔比星、盐酸柔红霉素、盐酸伊立替康、盐酸托泊替康及修饰后亲水的抗癌药物等。
进一步优选为,步骤(1)所述水溶性抗癌药为盐酸多柔比星、盐酸表柔比星、盐酸柔红霉素、盐酸伊立替康和盐酸托泊替康中的一种以上,最优选为dox。
优选的,步骤(1)所述水溶性抗癌药和sirna的摩尔比值为0.3~2。
优选的,步骤(1)所述sirna水溶液的浓度为6.0nmol/ml,体积为0.5ml,dox·hcl为0.5ml,配置不同的浓度。
优选的,步骤(1)所述室温孵育的时间为10-20min。
优选的,步骤(1)所述离心的转速为12000-15000g,离心的时间为5-10min。
由以上所述的制备方法制得的一种sirna和抗癌药物疏水性复合物。
以上所述的一种sirna和抗癌药物疏水性复合物应用于制备荷载hip[sirna&抗癌药物]纳米颗粒,用于共递送干扰核酸和水溶性抗癌药物,包括以下步骤:
将含有sirna和抗癌药物疏水性复合物和mpeg-b-pla的混合物溶解在二甲基亚砜中,然后在搅拌下加入水中,透析后,离心得到np[sirna&抗癌药物]。
优选的,所述sirna和抗癌药物疏水性复合物加入量为0.8mg,mpeg-b-pla的加入量为10.0mg,dmso用量为1.0ml,纯水用量为10ml。
优选的,所述搅拌的时间为2-4h;所述离心的转速为5000-8000rpm,时间为5-10min。
优选的,所述透析所用透析袋mwco=3500。
优选的,所述透析是在4℃下用超纯水透析4小时。
本发明中sirna是亲水性药物,抗癌药物也是亲水性的,通过hip策略制备的[sirna和抗癌药物]复合物是疏水性的,实现了药物从亲水到疏水的转变,通过疏水作用可以很容易包载到非阳离子载体peg-b-pla纳米颗粒中,然后递送到肿瘤细胞内协同发挥抗肿瘤效应。
与现有技术相比,本发明具有如下优点:
本发明通过hip策略制得的[sirna和抗癌药物]复合物实现了核酸药物和化疗药物由亲水性向疏水性的转变,可以很容易被包载到临床批准的(peg-b-pla)胶束中,且包封率高,可用于核酸药物和化疗药物的共递送,并且该方法可以扩展应用到其他具有疏水结构域的水溶性抗癌药物中,两种药物联用可以协同作用提高临床抗肿瘤效果,具有巨大的临床应用潜能。
附图说明
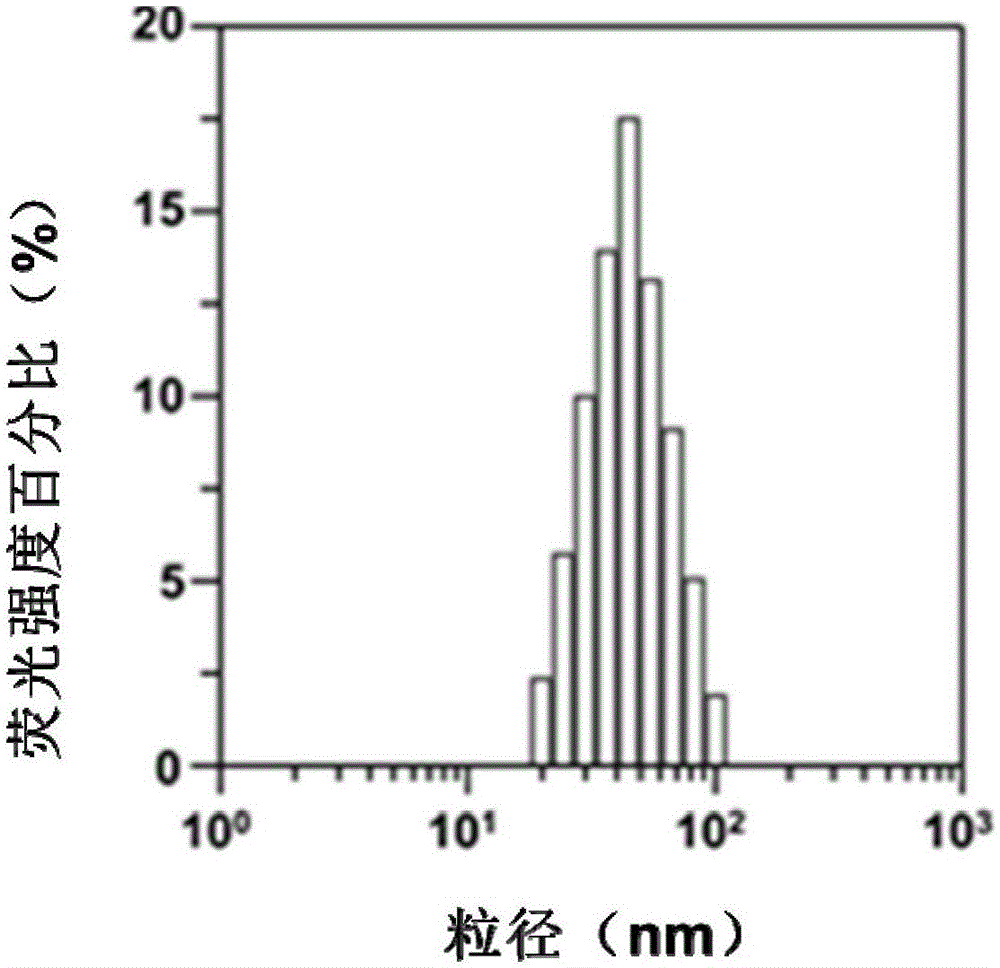
图1为包载hip[sirna和dox]的纳米颗粒的粒径分布图。
图2为包载hip[sirna和dox]的纳米颗粒的zeta电位图。
图3为包载hip[sirna和dox]的纳米颗粒的透射电子显微图片。
图4为dox在体外从纳米颗粒中的累积释放曲线图。
图5为用制备的纳米颗粒处理mda-mb-231细胞后,细胞中的plk1mrna的表达结果图。
图6为用制备的纳米颗粒处理mda-mb-231细胞后,细胞中的plk1蛋白的的表达结果图。
图7为在用不同制剂孵育后诱导mda-mb-231细胞凋亡情况图。
图8为静脉内给药后mda-mb-231异种移植小鼠肿瘤生长曲线图。
具体实施方式
以下结合实例对本发明的具体实施做进一步的说明,但本发明的实施方式不限于此。
实施例1、制备hip[sirna和抗癌药物(dox)]复合物
一、hip[sirna和dox]复合物的制备
将从大连美仑公司购买的dox·hcl(0.5ml)逐渐加入到从吉玛公司购买的fam-sirna(0.5ml,3.0nmol)的水溶液中,dox与sirna核苷酸(sirna-nt)单位摩尔比为1:1,在室温下温育10分钟后,将混合物溶液以12,000g的速度离心5分钟以收集形成的[sirna和dox]复合物的hip。其他水溶性抗癌药物和sirna疏水复合物制备方法与此相同。
实施例2、np[siplk1&dox]的制备及应用
一、np[siplk1&dox]的制备
np[siplk1&dox]通过纳米沉淀法制备,将含有sirna-dox疏水性复合物(0.8mg)和mpeg-b-pla(10.0mg)的混合物溶解在1.0ml二甲基亚砜(dmso)溶液中,然后在搅拌下加入10ml超纯水中,继续搅拌2小时后,将溶液转移到透析袋(mwco=3500)中,在4℃下用超纯水透析4小时,去掉有机溶剂dmso,然后将溶液以5000rpm的速度离心5分钟,得到含有np[sirna&dox]的上清液,将其储存在4℃下以备进一步使用。
二、np[siplk1&dox]的表征
hip[sirna&dox]复合物的包封效率约为41.16±0.47%,得到的np[sirna&dox]粒径约为53±5.7nm(见图1),ζ电位约为-13.2mv(见图2),类似于空白mpeg-b-pla纳米颗粒的ζ电位。另外,通过透射电子显微镜(tem)观察到的np[sirna&dox]呈致密的球形结构(见图3)。同时,得到的np[sirna和dox]在pbs和含有10wt%fbs的pbs中孵育80小时以上都没有显示出明显的粒径大小变化。通过测量dox的浓度来确定dox和sirna在ph7.4和ph5.5条件下从np[sirna和dox]中的释放曲线,如图4所示,在ph7.4和ph5.5条件下累计释放率分别为66.2%和88.6%。
三、np[siplk1&dox]的体外抑瘤实验
1、肿瘤细胞对np[siplk1&dox]的摄取和释放
用fam-sirna追踪np[sirna和dox]在细胞内的分布。将制备的np[fam-sirna&dox]与mda-mb-231细胞一起温育1小时,然后用pbs洗涤两次除去未内化的纳米颗粒,再孵育4小时和8小时后,通过流式细胞术分析确定细胞内fam-sirna和dox荧光。在共孵育2小时后可以观察到fam-sirna和dox荧光,在去除未内化的np[fam-sirna&dox]后,细胞内fam-sirna和dox荧光在进一步孵育另外4小时和8小时后显著升高。考虑到在形成fam-sirna和dox复合物后fam-sirna和dox荧光部分被淬灭,推测增强的fam-sirna和dox荧光应该归因于dox和sirna在肿瘤细胞内的解离。
2、np[siplk1&dox]在肿瘤细胞内的分布
利用激光共聚焦扫描显微镜观察fam-sirna和dox荧光在细胞内的分布。在孵育2小时后,可以观察到在细胞质中共定位的轻微fam-sirna和dox荧光。去除未内化的np[fam-sirna&dox]并进一步孵育4小时后,细胞内fam-sirna和dox荧光显著增强,并且在细胞核中观察到轻微的dox荧光。当孵育时间延长至8小时时,大多数dox荧光定位于细胞核中,而fam-sirna的荧光仍保留在细胞质中,这意味着sirna和dox在肿瘤细胞内完全解离。
3.np[siplk1&dox]在体外的抑瘤效果
实验选择在许多癌细胞中高表达的polo样激酶1(plk1)作为致癌靶基因,用含不同浓度sirna的np[siplk1和dox]转染mda-mb-231细胞,24h后通过qrt-pcr分析plk1基因的表达。如图5所示,负载阴性对照sirna(sin.c.)的np[sin.c.&dox]不影响plk1表达,而np[siplk1和dox]有效下调肿瘤细胞中plk1基因的表达,并且np[siplk1&dox]的基因沉默效率还具有剂量依赖性。例如,当siplk1浓度从20nm增加到40nm,60nm,80nm,100nm时,np[siplk1&dox]处理后plk1mrna水平显著降低至87.9%,78.9%,51.7%,30.3%和18.1%。此外通过蛋白质印迹分析48小时后plk1蛋白表达水平,如图6所示,用np[siplk1&dox]处理后在plk1蛋白表达水平也表现出剂量依赖性基因沉默现象,而用对照制剂np[sin.c.&dox]处理后未观察到plk1蛋白水平的显著降低,表明有效的plk1基因下调确实是由rnai效应诱导的。
据报道,plk1表达的下调诱导癌细胞凋亡。在与上述制剂一起温育48小时后,用膜联蛋白v-fitc和碘化丙锭(pi)染色mda-mb-231细胞以评估细胞凋亡。如图7所示,用含sin.c60nm和100nm的np[sin.c.和dox]转染mda-mb-231细胞时可分别诱导20.4%和32.1%的细胞凋亡,相应的dox浓度分别为2.52nm和4.2nm。相比之下,当用np[siplk1&dox]分别以60nm和100nm的siplk1剂量处理mda-mb-231细胞时,细胞凋亡显著升高至34.0%和46.5%。
四、np[siplk1&dox]的体内抗肿瘤治疗实验
每天向mda-mb-231异种移植的小鼠静脉内注射游离的siplk1,游离的dox,np[sin.c.&dox]或np[siplk1&dox],每次注射的sirna和dox的剂量分别为40μg和68.4μg。如图8所示,用游离siplk1处理几乎无抗癌功效,而用游离dox和np[sin.c.&dox]处理后轻微抑制肿瘤生长,这是由于dox产生了一定的治疗效果,np[siplk1&dox]组显示出最高的抗癌功效,这证实np[siplk1&dox]能够实现sirna疗法和化疗结合协同抑制肿瘤的效果。
- 还没有人留言评论。精彩留言会获得点赞!