一种利用倍增算法改进基于BWT变换的基因序列比对方法
wheeler transform)的预处理阶段进行改进。与传统的bwt中旋转构造矩阵并计算各种数据结构不同的是,改进后的方法直接通过reference参考长序列计算出后缀数组,并根据后缀数组快速计算出所需的数据结构。不仅可以在空间上不需要再存储bwt矩阵,省去了n*n(n为参考基因序列的长度)的二维空间,并且在时间上也获得较大的提升,因为不需要和原始bwt一样去对基因序列进行旋转、排序等耗时的操作。同时本发明也对reads种子序列进行了预处理操作,将reads种子序列排序后,使相同后缀的reads聚集在一起,这样方便保存上一个reads种子序列比对的中间结果(相同后缀),减少了比对次数。
14.与现有方法相比,本发明的有益效果主要表现在:
15.(1)通过倍增算法对reference参考长序列建立后缀数组sa,由于不需要建立传统bwt的n*n(n是reference的长度)矩阵,节省了空间以及时间复杂度。若按照传统的方式通过建立bwt矩阵,然后再对bwt矩阵排序得到sa后缀数组,时间和空间复杂度太大,因为reference的长度过于大,要对bwt矩阵的n行序列排序,时间复杂度最好也是o(n*n*logn)。而采用倍增算法直接对reference的后缀排序得到sa后缀数组的时间复杂度将会是o(n*logn),并且节省了n*n的空间。
16.(2)找到sa区间后利用bfs宽搜的方法进行搜索,宽搜可以保证最快找到最优的序列得分,不会漏掉最优的sa,然后用堆数据结构进行保存,每次堆顶都会是得分最高分,每次取得分最高项进行下一层搜索,一直到匹配结束就是最优的匹配结果。
17.(3)将具有相同后缀的reads序列聚集在一起,并保存比对中间结果,减少高速缓存命中率低的访问次数。
18.(4)通过实验证实,本发明利用倍增算法改进基于bwt变换的基因序列比对方法,在reference参考长序列较大时,通过倍增算法进行预处理,建立sa后缀数组处理时间有着非常大的提升。同时,本发明在精确比对的时间以及模糊比对的精度上,相较于现有比对方法软件,也存在一定程度的优势。
附图说明
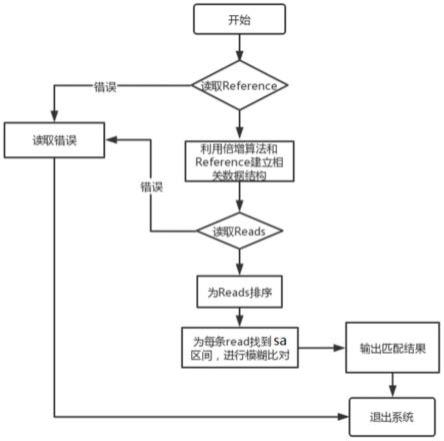
19.图1为本发明的算法流程图。
20.图2以reference:”acaacg$”($为终止字符)为例建立bwt矩阵,每行依次左移一位字符后并对bwt矩阵排序。
21.图3为bwt相关的数据结构(以reference:”acaacg”为例)。
22.图4为reads排序前后对比图。
23.图5为以reference:”acaacg”,read:”aac”为例的匹配过程。
具体实施方式
24.请参阅图1所示,本发明所提出的一种利用倍增算法改进基于bwt变换的基因序列比对方法,其步骤主要为:
25.步骤1:利用倍增算法对参考长序列reference计算出sa后缀数组,sa后缀数组记录n*n的bwt矩阵排序之前的行号,即f列的位置;其中n是参考长序列reference的长度;
26.步骤2:对参考长序列reference遍历建立c数组,c(a)数组计算在reference[0,n-2]比字符a小的元素个数,记录字符a的首地址;
[0027]
步骤3:利用sa后缀数组和参考长序列reference计算出l列,即n*n的bwt矩阵排序后最右边的一列;
[0028]
步骤4:通过建立好的c数组,得到f列,即n*n的bwt矩阵排序后最左边的一列;
[0029]
步骤5:利用步骤3的l列计算occ数组,occ(a,i)为a在l数组i+1行之前出现的次数,从而bwt所需要的数据结构建立完成(bwt相关数据结构如图3所示);
[0030]
步骤6:使用基数排序和计数排序相结合的方法对reads种子序列从右往左按照字典顺序排序,使得相同后缀的read聚集在一起;
[0031]
步骤7:通过比较上下两条read找出相同后缀,从右边起,第一个不相同的碱基就是比对的起始位置,确定read比对的起始位置之后,利用步骤1-5构建的bwt数据结构和bfs宽搜的方法找到sa区间,输出匹配的位置和错配的个数以及运行时间。
[0032]
本发明实现算法具体说明如下:
[0033]
1、利用倍增算法得到sa后缀数组(suffix array)
[0034]
以序列acaacg为例,它的后缀就是acaacg,caacg,aacg,acg,cg,g。后缀数组sa[i]就是排名为i的后缀在reference中起始位置的下标,实际记录排名为i的后缀在reference的实际位置,因为对bwt矩阵排序后位置是混乱的,用后缀数组记录实际位置。
[0035]
本发明所采用的倍增算法,主要的思路是:首先利用了两点性质,第一,后缀i和后缀j的前k个字符相等并且后缀i+k和后缀j+k的前k个字符相等。第二,后缀i的前2k个字符小于后缀j的前2k个字符。已知所有后缀前k个字符的大小关系就可以知道所有后缀的前2k个字符的大小关系。将后缀i的前k个字符的排名当做第一关键字,将后缀i+k的前k个字符的排名当做第二关键字,然后利用基数排序算出后缀i的前2k个字符的排名。
[0036]
下面以reference=“acaacg$”字符串为例,通过倍增算法构建后缀数组详细过程($比任何字符都小,表格中省略,排序时以0代替):
[0037]
表1倍增算法过程
[0038][0039]
通过表1可以看出,第一次排序,a最小标记为1,g最大标记为3,c次之为2。
[0040]
第一次倍增长度为1即第一列字符和第二列结合的后缀(ac即12),第二列字符和第三列结合的后缀(ca即21),一直到最后一列(g$即30)。然后对这6个两位数进行基数排序,不需要对全部的数进行基数排序,因为通过上一次的排序已经知道了第一个关键字(两位数当中十位数)的相同的个数以及排序位置,只需要对第一个关键字相同的数字进行排序,有x个第一关键字相同的数字,后面排序的数加(x-1)即可。
[0041]
第二次倍增长度为2即第一列和第三列结合的后缀(acaa即21),第二列和第四列结合的后缀(caac即32),一直到最后一列(g$即50),然后对再次基数排序(第三次排序)。
[0042]
如果排序结果得到的数字各不相同则排序结束,因为再倍增下去,得到的两位数中的十位数都是上一次排序的结果。例如表1中第三次排序的结果没有相同数字,再倍增下去,得到的第四次排序结果还是2,4,1,3,5,6。
[0043]
每次将排序长度*2,即持续2倍倍增,最多需要log(n)次便可以完成排序,而传统的对bwt矩阵排序则需要n(n是reference的长度)次排序。
[0044]
将得到的排序数组记为rank[i]数组,下标i代表关键字位置,值表示关键字大小,简单来说就是sa后缀数组表示排第几的是谁,而rank则表示“你排第几”。
[0045]
以reference=“acaacg$”为例,经过倍增算法后得到rank[i]=2,4,1,3,5,6。
[0046]
rank[0]=2表示sa后缀数组的第2位索引的关键字是0。
[0047]
rank[1]=4表示sa后缀数组的第4位索引的关键字是1。
[0048]
rank[2]=1表示sa后缀数组的第1位索引的关键字是2。
[0049]
rank[3]=3表示sa后缀数组的第3位索引的关键字是3。
[0050]
rank[4]=5表示sa后缀数组的第5位索引的关键字是4。
[0051]
rank[5]=6表示sa后缀数组的第6位索引的关键字是5。
[0052]
由图2可以看出,传统构建矩阵方法中,经过左移后矩阵的最后一行(第6行,从0开始)的开头一定是终止字符$,又因为$最小,所以对矩阵排序后第0行开头字符一定是$,即sa[0]=6(即reference的长度)。由次可得到sa[i]=6,2,0,3,1,4,5。由此可见,本发明不通过传统建立bwt矩阵的方法也能得到sa后缀数组。
[0053]
2、利用reference和sa后缀数组得到bwt变换(burrows-wheeler transform)剩余所有的数据结构。
[0054]
c(a)数组是计算reference序列中比a小的字符个数,大小是$《a《c《g《t,c(a)的实际意义其实是为了计算字符a的首地址,c(a)+1就是a字符的首地址。因为c(a)是计算比a小的,当遇到字符a的时候,c,g,t对应的c(c),c(g),c(t)都是比c(a)大,因为c,g,t都比a大,所以c,g,t对应的c(c),c(g),c(t)都要加1。这个时间复杂度也就是o(n),n是指reference的长度。
[0055]
f数组表示bwt矩阵排序后的第一列,所以相同字符都是聚在一起的,通过上述建立好的c数组可以直接得到f数组,f[0]=
‘
$’,f[1]=’a’,f[x]=
‘
c’,x的值为c[c],f[y]=
‘
g’,y的值为c[g],依次算出f数组。
[0056]
从图3的bwt矩阵中的关系可以看出,f列和l列一定在原基因序列中相邻。由于sa后缀数组表示f数组的位置,而l数组又是f数组的前一个字符,只需要直接根据sa的位置来对原基因序列截取即可。
[0057]
l数组建立完成后,通过l数组建立occ数组,occ(a,i)是一个二维数组,表示a字符在i+1行的l列之前出现的次数,以图3中occ(a,5)=3为例,其表示在l列中第6行之前出现过3次。
[0058]
3、对多个reads种子序列排序
[0059]
本发明采用基数排序和计数排序相结合的算法对reads进行排序,此算法针对reads排序效率较高。
[0060]
首先第一步,建立reads数据的哈希索引:读取reads文件,产生正反两向reads,并统计相同哈希键值出现的数目,建立reads数据的哈希索引。
[0061]
第二步,依据reads哈希索引,分别载入相应的reads数据块,之后利用基数排序算法与计数排序算法对reads数据块进行排序。对同一哈希块内的reads(即前k-m相同的一组reads),排除k-m后,分成8bp长度的若干段,进行基数排序(如read长度为48bp,k=16时,剩余的32bp,分成4组,每组长度为8bp)。每次排序8bp时,采用计数排序。
[0062]
计数排序过程中,有时数据块很大,例如几百万条数据以上,有时数据块很小,仅几十条,这导致了有时数据密集,有时数据稀疏。数据密集的情况,本发明采用了上述类似k-m哈希分类排序的方法。数据稀疏时,将8bp又分成2组每组4bp,进行基数排序。数据量小于64bp时,直接采用快速排序即可。
[0063]
reads数据排序时间复杂度为o(l*k),l是reads的长度,k是reads数据块的大小。reads排完序后有一个特点,就是后缀相同的reads会聚到一起,如图4所示。这样就使得相似度大的reads聚在一起,减少比较次数。
[0064]
4、找到sa区间并进行序列比对
[0065]
bwt相关的数据结构建立完成后,利用c数组找到sa后缀数组的第一轮的起始和结尾位置。用sp表示sa区间的起始位置,ep表示sa区间的结尾。比对第一个read序列从read最
后一位从右往左比对,并和下一条read比较,得到相同的后缀区间,保存相同后缀比对的结果(sa区间)。对于下一条read的比对,它的比对起始位置是read的长度减去和上一条read的相同后缀区间,也就是从右往左第一个不相同的碱基就是read比对的起始位置。
[0066]
第一轮sp和ep(i表示种子序列比对的起始位置):
[0067]
sp=c[i]
[0068]
ep=c[i+1]-1
[0069]
然后利用backward-search算法公式计算之后的sa区间,并用堆的数据结构进行保存。backward-search算法公式如下:
[0070]
sp(iq)=c[i]+occ[i,sp(q)-1]
[0071]
ep(iq)=c[i]+occ[i,ep(q)]-1
[0072]
图5更加清楚的展示这个匹配过程,以reference=”acaacg”,read=”aac”为例。
[0073]
用backward-search算法公式找到匹配的sa区间,在sa区间寻找匹配的序列,再用堆的数据结构加宽搜bfs找到可以插入空格,允许错配的差异。利用bfs宽搜的方法可以保证最快找到最优的序列得分。
[0074]
其中,宽搜的步骤如下:
[0075]
步骤1:初始化建立一个优先队列substrheap,初试序列为空,即得分初试为0.
[0076]
步骤2:初始化一个heap变量(初试为0),保存每次搜索的结果,并对每次的结果打分(打分标准为插入空格和错配的次数越少得分越高),堆顶为分数最高项。
[0077]
步骤3:利用backward-search算法公式计算sa区间,然后判断此read是否为reference的子串,如果不是则插入空格或者错配进行再次匹配,如果超出误差上界则匹配失败。在误差内匹配成功则用堆保存中间结果,继续匹配,每次取得分最高项进行下一层搜索。
[0078]
步骤4:对mismatch,gap open,match等进行判断然后进行打分,然后堆数据结构进行保存,每次堆顶都会是得分最高分,就是最优的匹配分数,最终输出也是利用堆保存的数据进行输出。
[0079]
实施例
[0080]
实施例所用的参考基因组节选自威廉姆斯公司82号大豆栽培品种的一号染色体,基因组数据文件从美国国家信息中心官方网站下载所得(national center for biotechnology information,http://www.ncbi.nlm.nih.gov)。编号为》nc_016088.4glycine max cultivar williams 82chromosome 1,glycine_max_v4.0,whole genome shotgun sequence。节选长度为50000bp。对比基因节选自豌豆的基因组组装的1lg6染色体。编号为》lr535892.1pisum sativum genome assembly,chromosome:1lg6,whole genome shotgun sequence,每一段短序列的节选长度为100bp。
[0081]
1、是否采用倍增算法建立sa后缀数组的时间比较(仅在对reference的预处理阶段,与传统bwt比较):
[0082]
表2是否采用倍增的时间比对
[0083][0084]
从表2中的结果可以看出在reference较大时,通过倍增算法进行预处理(建立sa后缀数组)时间有非常大的提升,传统bwt预处理时间非常慢。
[0085]
2、精确比对的时间比较:
[0086]
表3三种软件时间比对结果
[0087][0088]
从三种方法软件的比对时间来说,本发明在较低数量read的比对时间明显优于bowtie2、bwa两种软件,但是在加大read条数的时候,所表现出来的结果不是最好的,低于bwa软件,但高于bowtie2软件。
[0089]
3、模糊比对的精度比较:
[0090]
表4三种软件精度比对结果
[0091][0092][0093]
从三种方法软件的比对精度来说,本发明整体表现不错,特别是在较低数量read条件下精度很高,但是随着条数增加,对比精度略有下降,说明bfs宽搜的方式虽然会加快搜索,但是仍然会丢失一部分精度。
[0094]
总体来说,本发明利用倍增算法改进基于bwt变换的基因序列比对方法,在reference参考长序列较大时,通过倍增算法进行预处理,建立sa后缀数组处理时间有着非常大的提升。同时,本发明在精确比对的时间以及模糊比对的精度上,相较于现有比对方法软件,也存在一定程度的优势。
[0095]
以上内容仅仅是对本发明的构思所作的举例和说明,所属本技术领域的技术人员对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,只要不偏离发明的构思或者超越本权利要求书所定义的范围,均应属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1