静电荷图像显影剂、处理盒、图像形成装置和图像形成方法与流程
本发明涉及静电荷图像显影剂、处理盒、图像形成装置和图像形成方法。
背景技术:
JP-A-2009-156902(专利文献1)公开了一种在下述装置中进行的高速的全色图像形成方法,所述装置包含:至少承载色调剂图像的电子照相感光体、对电子照相感光体进行充电的充电单元、在电子照相感光体上形成静电潜像的曝光单元、通过色调剂使电子照相感光体上形成的静电潜像显影以在电子照相感光体上形成色调剂图像的显影单元、将电子照相感光体上形成的色调剂图像转印到中间转印介质上的一次转印单元、将中间转印介质上的色调剂图像转印到转印材料上的二次转印单元、通过热压定影部件将色调剂图像定影在转印材料上的定影单元以及清洁转印后残留在电子照相感光体表面上的色调剂的清洁单元,其中,电子照相感光体的线速度为300mm/秒~1000mm/秒,二次转印咬合部的转印时间为0.5毫秒~20毫秒,其中,色调剂至少包含树脂和着色剂,色调剂颗粒的重均粒径为1μm~6μm,一次平均粒径为80nm~500nm的微粒附着于色调剂的表面上,相对于100重量%的色调剂,一次平均粒径为80nm~500nm的微粒的添加量为0.5重量%~5重量%,色调剂1的颗粒表面通过纳米压痕法测定的硬度为1GPa~3GPa,色调剂1的颗粒表面通过微米压痕法测定的硬度为40N/mm2~120N/mm2。
技术实现要素:
本发明的目的在于提供一种静电荷图像显影剂,其中抑制了色调剂的充电量的降低。
根据本发明的第一方面,提供一种静电荷图像显影剂,所述静电荷图像显影剂包含:静电荷图像显影用色调剂,所述静电荷图像显影用色调剂包含色调剂颗粒和外添剂,所述色调剂颗粒包含具有松香骨架的聚酯树脂;和载体,所述载体具有芯材颗粒和涂覆所述芯材颗粒的表面的涂覆树脂层,其中,所述涂覆树脂层的硬度为所述色调剂颗粒的硬度的1.2倍~2.0倍。
根据本发明的第二方面,在第一方面所述的静电荷图像显影剂中,所述涂覆树脂层的树脂可以包含选自由下述物质组成的组中的至少一种物质作为聚合成分:(甲基)丙烯酸二环戊基酯、(甲基)丙烯酸异冰片基酯、(甲基)丙烯酸金刚烷基酯、(甲基)丙烯酸降冰片基酯和(甲基)丙烯酸羟基萘基酯。
根据本发明的第三方面,提供一种处理盒,所述处理盒包含显影单元,所述显影单元容纳有第一方面或第二方面所述的静电荷图像显影剂,并通过所述静电荷图像显影剂使图像保持体的表面上形成的静电荷图像显影从而形成色调剂图像,并且所述处理盒能在图像形成装置上安装和拆卸。
根据本发明的第四方面,提供一种图像形成装置,所述图像形成装置包含:图像保持体;充电单元,所述充电单元对所述图像保持体的表面进行充电;静电荷图像形成单元,所述静电荷图像形成单元在所述图像保持体的表面上形成静电荷图像;显影单元,所述显影单元容纳有第一方面或第二方面所述的静电荷图像显影剂,并通过所述静电荷图像显影剂使所述静电荷图像显影,从而形成色调剂图像;转印单元,所述转印单元将所述色调剂图像转印到记录介质上;和定影单元,所述定影单元将所述色调剂图像定影在所述记录介质上。
根据本发明的第五方面,提供一种图像形成方法,所述图像形成方法包括:对图像保持体的表面进行充电;在所述图像保持体的表面上形成静电荷图像;通过第一方面或第二方面所述的静电荷图像显影剂使所述静电荷图像显影,从而形成色调剂图像;将所述色调剂图像转印到记录介质上;和将所述色调剂图像定影在所述记录介质上。
根据本发明的第一方面,可以获得一种静电荷图像显影剂,所述静电荷图像显影剂包含静电荷图像显影用色调剂和载体,所述静电荷图像显影用色调剂包含色调剂颗粒和外添剂,所述色调剂颗粒包含具有松香骨架的聚酯树脂,所述载体具有芯材颗粒和涂覆所述芯材颗粒的表面的涂覆树脂层,其中,与涂覆树脂层的树脂的硬度在色调剂颗粒硬度的1.2倍~2.0倍范围之外的情况相比,色调剂的充电量降低得到抑制。
根据本发明的第二方面,可以获得一种静电荷图像显影剂,其中,与涂覆树脂层的树脂包含除选自(甲基)丙烯酸二环戊基酯、(甲基)丙烯酸异冰片基酯、(甲基)丙烯酸金刚烷基酯、(甲基)丙烯酸降冰片基酯和(甲基)丙烯酸羟基萘基酯的至少一种之外的其他物质作为聚合成分的情况相比,涂覆树脂层的树脂可抑制载体中的涂覆树脂层剥落。
根据本发明的第三方面至第五方面,可以获得一种处理盒、图像形成装置和图像形成方法,与施用下述静电荷图像显影剂(所述静电荷图像显影剂包含静电荷图像显影用色调剂和载体,所述静电荷图像显影用色调剂包含色调剂颗粒和外添剂,所述色调剂颗粒包含具有松香骨架的聚酯树脂,所述载体具有芯材颗粒和涂覆所述芯材颗粒表面的涂覆树脂层,涂覆树脂层的树脂的硬度在色调剂颗粒硬度的1.2倍~2.0倍范围之外)的情况相比,所述处理盒、图像形成装置和图像形成方法各自均可提供由静电荷图像显影用色调剂的充电量降低导致的图像缺陷得到抑制的图像。
附图说明
将基于以下附图对本发明的示例性实施方式进行详细描述,在附图中:
图1是显示本示例性实施方式的图像形成装置的实例的示意性构造图;和
图2是显示本示例性实施方式的处理盒的实例的示意性构造图。
具体实施方式
下文将对作为本发明实例的示例性实施方式进行详细描述。
[静电荷图像显影剂]
本示例性实施方式的静电荷图像显影剂(下文某些情况中称之为“显影剂”)包含静电荷图像显影用色调剂(下文某些情况中称之为“色调剂”)和载体,所述静电荷图像显影用色调剂包括含有色调剂颗粒和外添剂的色调剂,所述色调剂颗粒包含具有松香骨架的聚酯树脂(下文某些情况中称之为“特定聚酯树脂”),所述载体具有芯材颗粒和涂覆所述芯材颗粒表面的涂覆树脂层,其中,所述涂覆树脂层的硬度为所述色调剂颗粒的硬度的1.2倍~2.0倍。
此处,由于松香所具有的特性,含特定聚酯树脂的色调剂颗粒倾向于比采用其中不具有松香骨架的树脂的色调剂颗粒具有更高的硬度。
此外,具有高硬度的这类色调剂颗粒倾向于使色调剂中的外添剂不易埋入色调剂颗粒中。
即,包含这种含有外添剂和具有高硬度的色调剂颗粒的色调剂的显影剂可抑制因外添剂埋入色调剂颗粒中所致的色调剂充电量的降低,因此,据认为当将所述显影剂应用于图像形成时,可以抑制因色调剂飞散所致的图像缺陷的产生或起脏(印刷物的非图像部分的污染)。
另一方面,据认为包含上述色调剂的显影剂具有外添剂不易埋入色调剂颗粒中但容易埋入载体的涂覆树脂层中的倾向。
基于此原因,当采用包含上述色调剂的显影剂形成图像时,外添剂凭借图像形成中的振动而埋入载体的涂覆树脂层中,因此,降低了在载体中向色调剂提供电荷的能力,结果,具有色调剂的充电量降低的趋势。
因此,当本示例性实施方式的显影剂具有上述构成时,可以抑制色调剂充电量的降低。
其原因未明,但据推测如下。
本示例性实施方式的显影剂包含色调剂和具有涂覆树脂层的载体,所述色调剂含有具有高硬度的色调剂颗粒,所述涂覆树脂层的硬度高于所述色调剂颗粒的硬度。
因此,例如,即使在进行其中通过提供振动的TURBULA混合机或调节供应至显影套筒的显影剂量的调节部件而施加冲击的图像形成步骤的情况中,也具有外添剂不易埋入色调剂颗粒和载体任一种中的趋势。
基于此原因,可以抑制因外添剂埋入色调剂颗粒中所致的色调剂充电量的降低,并且可以抑制因外添剂埋入载体的涂覆树脂层中由此降低载体提供电荷的能力而导致的色调剂充电量的降低。因此,使用本示例性实施方式的显影剂,可以从色调剂和载体两方面抑制图像形成中所涉及的色调剂充电量的降低。
由上可见,使用本示例性实施方式的显影剂,可以抑制色调剂充电量的降低。
而且,即使在连续图像形成中,在使用本示例性实施方式的显影剂时,也可以抑制色调剂充电量的降低,结果,例如,抑制了因色调剂充电量的降低所致的因色调剂飞散导致的图像缺陷的产生或背景沾污(印刷物的非图像部分的污染)。
此外,据认为,即使在连续图像形成中,在使用本示例性实施方式的显影剂时,也可以抑制色调剂充电量的降低,结果,增加了使用寿命。
本文中,涂覆树脂层的硬度与色调剂颗粒的硬度之比(涂覆树脂层的硬度/色调剂颗粒的硬度)为1.2~2,优选为1.5~1.85,更优选为1.6~1.8。
当涂覆树脂层的硬度与色调剂颗粒的硬度之比为1.2以上时,抑制了外添剂埋入载体的涂覆树脂层中,因此,抑制了载体提供电荷的能力的下降。
此外,当涂覆树脂层的硬度与色调剂颗粒的硬度之比等于或小于2.5时,与色调剂颗粒的硬度相比,载体的涂覆树脂层的硬度显著增加,因此,抑制了外添剂不易埋入而外添剂仅易于埋入色调剂颗粒中的现象,结果,抑制了色调剂充电量的降低。
而且,涂覆树脂层的硬度为0.168GPa~0.28GPa,优选为0.21GPa~0.259GPa,更优选为0.224GPa~0.252GPa。
此外,色调剂颗粒的硬度为0.1GPa~0.2GPa,优选为0.12GPa~0.18GPa,更优选为0.14GPa~0.16GPa。
色调剂颗粒的硬度和涂覆树脂层的硬度通过下述方法确定。
涂覆树脂层的硬度通过测量如下制备获得的测量用树脂层而确定。
首先,从显影剂中回收载体,将载体表面上的涂覆树脂层浸渍在四氢呋喃溶液中,并将溶出的树脂层和沉降的芯材颗粒彼此分离并剥落。
收集由此剥落的涂覆树脂层并用作样品。
接下来,将从膜厚为300μm的硅晶片上切下的10mm×10mm的基片放在铝盘上,并将通过将样品溶解在四氢呋喃溶液中至10重量%的浓度所形成的溶液倒入其中。确定倒入的溶液量以使溶剂蒸发后基片上的树脂层的厚度为30μm~40μm。所述溶液在铝盘上于常温(例如25℃)干燥,然后通过具有加热功能的设备在70℃的温度继续干燥5小时,从而获得厚度为30μm~40μm的测量用树脂层。
色调剂颗粒的硬度通过测量以下述方式制备并获得的测量用片剂来确定。
首先,从显影剂中回收色调剂,将其分散在离子交换水中,以超声波进行照射,从而分离外添剂和色调剂颗粒,然后进行过滤和洗涤处理,从而仅回收色调剂颗粒。
收集由此回收的色调剂颗粒并以直径为12mm的压片机施加60KN的载荷,从而获得高度为8mm、直径为12mm的测量用片剂。
对于测量用树脂层和测量用片剂,采用Nanoindenter(注册商标,由MTS SystemsInc.制造)测量硬度。
在下述条件下在同一测量用树脂层和同一测量用片剂中的十个点进行测量:最大载荷(Pmax)0.8[mN],所用压头:金刚石,三棱锥的Berkovich型,温度:23℃。
具体而言,在测量用树脂层的情况中,对于硅晶片上的测量用树脂层的表面,从任意位置开始在给定的方向上以1mm的间隔在五个点测量硬度,并进一步在与给定方向垂直的方向上以1mm的间隔在五个点测量硬度,而在测量用片剂的情况中,对于其表面,从任意位置开始在给定的方向上以1mm的间隔在五个点测量硬度,同样地,对于背面在五个点处测量硬度。
采用下式确定各硬度值并取平均值。
式:H[GPa]=Pmax[N]/A[mm2]
其中,H代表硬度,Pmax代表最大载荷,A代表压头附着到的印痕的投影截面积。硬度可以以动态硬度和肖氏载荷这两种形式获得,但除非特别指出,否则使用的是肖氏硬度。
下面,将详细描述本示例性实施方式的色调剂用聚酯树脂。
<静电荷图像显影用色调剂>
静电荷图像显影用色调剂包含含有特定聚酯树脂的色调剂颗粒和外添剂。
(色调剂颗粒)
色调剂颗粒包含粘合剂树脂,如果需要,还包含着色剂、防粘剂和其他添加剂。
粘合剂树脂的实例包括非晶性树脂,作为非晶性树脂,也可采用上述特定聚酯树脂,但也可以将其与特定聚酯树脂以外的其他非晶性树脂组合使用。
此外,作为粘合剂树脂,可以将结晶性树脂与非晶性树脂组合使用。
然而,相对于100重量份的全部粘合剂树脂,特定聚酯树脂的含量优选为70重量份以上,更优选为90重量份以上。
另外,非晶性树脂是指在采用差示扫描量热仪(DSC)的热分析测量中不具有明确的吸热峰而仅具有阶梯式吸热变化的树脂,其在常温(例如25℃)为固体并且在不低于玻璃化转变温度的温度热塑化。
相反,结晶性树脂是指在采用差示扫描量热仪(DSC)的测量中不具有阶梯式吸热变化而具有明确的吸热峰的树脂。
具体而言,例如,结晶性树脂是指在加热速率为10℃/分钟的测量中,吸热峰的半峰宽等于或低于10℃的树脂,非晶性树脂是指半峰宽超过10℃但没有观察到明确的吸热峰的树脂。
首先,将描述特定聚酯树脂。
特定聚酯树脂是具有松香骨架的聚酯树脂,其实例包括下示缩聚物的构成。
其中,从充电性的角度考虑,有利的是其中采用具有松香的单体作为多元醇的2)中所示的缩聚物。
1)由包括具有松香的多元羧酸的多元羧酸成分以及如果需要的包括其他多元羧酸的多元羧酸成分,与包括其他多元醇的多元醇成分形成的缩聚物。
2)由包括其他多元羧酸的多元羧酸成分,与包括具有松香的多元醇和如果需要的其他多元醇的多元醇成分形成的缩聚物。
3)由包括具有松香的多元羧酸和如果需要的其他多元羧酸的多元羧酸成分,与包括具有松香的多元醇和如果需要的其他多元醇的多元醇成分形成的缩聚物。
4)由松香、包括其他多元羧酸的多元羧酸成分与包括其他多元醇的多元醇成分形成的缩聚物。
即,具有松香的单体的实例包括松香(改性前松香)、具有松香的多元羧酸和具有松香的多元醇。通过使用这些单体作为缩聚物成分,使得特定聚酯树脂具有松香(松香骨架)。
不过,特定聚酯树脂中包含的源自松香的骨架的含量(下文在某些情况中称之为松香骨架的含量)为,例如,等于或小于70重量%,优选为5重量%~70重量%,更优选为10重量%~45重量%。
据认为,当该含量在上述范围内时,抑制了外添剂的埋入,并且容易实现对色调剂充电量降低的抑制。
通过特定聚酯树脂合成中包含松香的单体的种类和用量来调节松香骨架的含量。
本文中,松香骨架的含量是指由包含松香的单体作为缩聚成分的缩聚反应所形成的特定聚酯树脂中存在的松香骨架(即,结合到特定聚酯树脂的松香)的比例。
松香骨架的含量如下测量:采用NMR、LC-MS或GC-MS等分离特定聚酯树脂的成分,然后将其定量,由此测量在树脂中的含量。
此外,其他多元羧酸是除具有松香的多元羧酸以外的羧酸,其他多元醇是除具有松香的多元醇以外的多元醇。
松香
松香是由乔木和灌木获得的树脂酸的统称,其为由包含松香酸(作为三环二萜之一)及其异构体作为主要成分的源自天然产物的材料。松香的成分的具体实例除了松香酸外,还包括长叶松酸、新松香酸、海松酸、脱氢松香酸、异海松酸和山达海松酸,本示例性实施方式中所用的松香是这些酸的混合物。
松香大体分为由木浆作为原料获得的妥尔松香(tall rosin),由生松焦油作为原料获得的脂松香(gum rosin),以及由松树残干作为原料获得的木松香。
作为松香,优选的是脂松香和妥尔松香中的至少一种,因为其易于获得。
松香优选为精制松香。精制松香是被认为由来自未精制松香的树脂酸的过氧化物产生的高分子物质,或者通过除去未精制松香中所含的不可皂化物所获得的物质。
精制方法没有特别限制,可选自各种已知精制方法。其具体实例包括蒸馏、重结晶和提取。从工业角度考虑,优选的是通过蒸馏进行精制。考虑到蒸馏时间,通常选择在200℃~300℃的温度和等于或小于6.67kPa的压力进行蒸馏。重结晶例如通过下述方式进行:将未精制松香溶解在良溶剂中,然后蒸发溶剂以提供浓缩溶液,并向溶液中添加不良溶剂。良溶剂的实例包括:如苯、甲苯和二甲苯等芳香烃类;如氯仿等氯代烃类;如低级醇等醇类;如丙酮等酮类;以及如乙酸乙酯等乙酸酯类;不良溶剂的实例包括:如正己烷、正庚烷、环己烷和异辛烷等烃类溶剂。提取是包括下述步骤的方法:采用例如碱水获得未精制松香的碱性水溶液,采用有机溶剂提取溶液中所含的不溶的不可皂化物,然后中和水层以获得精制松香。
松香可以是歧化松香。歧化松香是脱氢松香酸和二氢松香酸作为主要成分的混合物,其中通过在歧化催化剂的存在下于高温加热包含松香酸作为主要成分的松香,从而除去分子中的不稳定共轭双键。
歧化催化剂的实例包括各种已知的歧化催化剂,包括如钯碳、铑碳和铂碳等负载催化剂,如镍和铂的粉末等金属粉末,碘,以及如碘化亚铁等碘化物。
为了除去分子中的不稳定共轭双键,松香可以是氢化松香。对于氢化反应,选择用于氢化反应的已知条件。即,通过在氢化催化剂的存在下在氢气加压下加热松香而进行反应。氢化催化剂的实例包括各种已知的氢化催化剂,包括如钯碳、铑碳和铂碳等负载催化剂,如镍和铂的粉末等金属粉末,碘,以及如碘化亚铁等碘化物。
此外,歧化松香和氢化松香可以在歧化处理或氢化处理之前或之后包括上述精制步骤。
多元羧酸成分
具有松香的多元羧酸(下文在某些情况中称之为羧酸改性松香)是通过使松香与α,β-不饱和羧酸(例如,α,β-不饱和羧酸或其酸酐)反应所形成的羧酸,其具体实例包括以羧酸(例如,(甲基)丙烯酸、富马酸、马来酸、马来酸酐、衣康酸和柠康酸(柠康酸酐))改性的松香。
羧酸改性松香的典型实例包括(甲基)丙烯酸改性松香、富马酸改性松香和马来酸改性松香。
此外,作为改性前的松香,可以采用与上述用作特定聚酯树脂的缩聚成分的松香相同的松香。
(甲基)丙烯酸改性松香是以(甲基)丙烯酸改性的松香。
(甲基)丙烯酸改性松香的具体实例包括通过使改性前松香与(甲基)丙烯酸进行加成反应所获得的改性松香,其更具体的实例包括通过改性前松香的主要成分中具有共轭双键的酸与(甲基)丙烯酸之间在加热时进行Diels-Alder反应所获得的改性松香。
作为(甲基)丙烯酸改性松香,从Diels-Alder反应中的反应活性的角度考虑,优选的是通过以丙烯酸改性所形成的具有低位阻的丙烯酸改性松香。
此外,“(甲基)丙烯酰基”是指丙烯酰基或甲基丙烯酰基。即,“(甲基)丙烯酸”是丙烯酸或甲基丙烯酸。并且,“(甲基)丙烯酸改性松香”是指以丙烯酸改性的松香或以甲基丙烯酸基改性的松香。
在(甲基)丙烯酸改性松香中,从增加聚酯的分子量和减少低分子量低聚物成分的角度考虑,(甲基)丙烯酸对松香的改性度(下文在某些情况中称之为(甲基)丙烯酸改性度)优选为5~105,更优选为20~105,进而更优选为40~105,进而更优选为60~105。
(甲基)丙烯酸改性度Xa通过下式(Aa)计算,其中式(Aa)的值越大,改性度越高。
式(Aa):Xa=[(Xa1-Y)/(Xa2-Y)]×100
在式(Aa)中,Xa1表示计算改性度的(甲基)丙烯酸改性松香的SP值,Xa2表示通过使1摩尔(甲基)丙烯酸与1摩尔松香反应可获得的(甲基)丙烯酸改性松香的饱和SP值,Y表示松香的SP值。
本文中,SP值是指以环和球式自动软化点测试仪确定的软化点。具体而言,SP值是通过下述步骤获得的值:将熔融状态的所需样品注入环,然后将样品冷却至室温(例如25℃),而后基于JIS B7410在下述条件下进行测试。
测试装置:环球式自动软化点测试仪ASP-MGK2(通过MEITECH Co.,Ltd.制造)
加热速率:5℃/分钟
加热开始温度:40℃
测量溶剂:甘油
此外,饱和SP值是:当(甲基)丙烯酸与松香之间进行反应直至达到所获得的(甲基)丙烯酸改性松香的SP值的饱和值时的SP值。
制备(甲基)丙烯酸改性松香的方法没有特别限制,但例如可以通过下述步骤获得(甲基)丙烯酸改性松香:将改性前松香与(甲基)丙烯酸混合,并将混合物加热至180℃~260℃的温度(优选为180℃~210℃)以进行Diels-Alder反应,从而将(甲基)丙烯酸加成至松香所包含的含共轭双键的酸。
反应后,(甲基)丙烯酸改性松香可原样使用,也可以通过如蒸馏等工序进一步精制而后使用。
富马酸改性松香是以富马酸改性的松香。
富马酸改性松香的具体实例包括通过使改性前松香与富马酸进行加成反应所获得的改性松香,其更具体的实例包括通过改性前松香的主要成分中具有共轭双键的酸与富马酸之间在加热时进行Diels-Alder反应所获得的改性松香。
在富马酸改性松香中,从增加聚酯的分子量和提高玻璃化转变温度的角度考虑,富马酸对松香的改性度(下文在某些情况中称之为富马酸改性度)优选为5~105,更优选为20~105,进而更优选为40~105,再进而更优选为60~105。
富马酸改性度Xf通过下式(Af)计算,其中式(Af)的值越大,改性度越高。
式(Af):Xf=[(Xf1-Y)/(Xf2-Y)]×100
其中,Xf1表示计算改性度的富马酸改性松香的SP值,Xf2表示通过使1摩尔富马酸与0.7摩尔松香反应可获得的富马酸改性松香的SP值,Y表示松香的SP值。
本文中,SP值是指以环球式自动软化点测试仪确定的软化点,具体而言,是通过上述方法测量的值。
制备富马酸改性松香的方法没有特别限制,但例如,富马酸改性松香可以通过下述步骤获得:将松香与富马酸混合,并将混合物加热至180℃~260℃的温度(优选为180℃~210℃)以进行Diels-Alder反应,从而将富马酸加成至松香所包含的含共轭双键的酸。
反应后,富马酸改性松香可原样使用,也可以通过如蒸馏等工序进一步精制而后使用。
在制备富马酸改性松香的方法中,优选的是使松香和富马酸在苯酚类的存在下反应,通过该化合物,松香与富马酸之间的反应效率易于提高。
所述苯酚类优选为二元酚和在羟基的邻位具有至少一个取代基的苯酚化合物(下文在某些情况中称之为受阻酚),更优选的是受阻酚。
二元酚是指其中有两个OH基团结合到苯环但其他取代基没有结合到苯环的化合物,具体而言,例如优选的是对苯二酚。受阻酚的具体优选实例包括叔丁基邻苯二酚。
相对于100重量份的富马酸改性松香的原料单体,苯酚类的用量优选为0.001重量份~0.5重量份,更优选为0.003重量份~0.1重量份,进而更优选为0.005重量份~0.1重量份。
马来酸改性松香是以马来酸或马来酸酐改性的松香。
马来酸改性松香的具体实例包括通过使改性前松香与马来酸或马来酸酐进行加成反应所获得的改性松香,其更具体的实例包括通过改性前松香的主要成分中具有共轭双键的酸与马来酸或马来酸酐之间在加热时进行Diels-Alder反应所获得的改性松香。
从增加聚酯的分子量和减少低分子量低聚物成分的角度考虑,松香的马来酸或马来酸酐改性度(下文在某些情况中称之为马来酸改性度)优选为30~105,更优选为40~105,进而更优选为50~105,进而更优选为60~105,特别优选为70~105。
马来酸改性度Xm通过下式(Am)计算,其中式(Am)的值越大,改性度越高。
式(Am):Xm=[(Xm1-Y)/(Xm2-Y)]×100
在式(Am)中,Xm1表示计算改性度的马来酸改性松香的SP值,Xm2表示通过使1摩尔马来酸与1摩尔松香在230℃反应可获得的马来酸改性松香的SP值,Y表示松香的饱和SP值。
本文中,SP值是指以环球式自动软化点测试仪确定的软化点,具体而言,是通过上述方法测量的值。
此外,饱和SP值是:当马来酸与松香之间进行反应直至达到所获得的马来酸改性松香的SP值的饱和值时的SP值。
制备马来酸改性松香的方法没有特别限制,但例如可以通过下述步骤获得马来酸改性松香:将改性前松香与马来酸或马来酸酐混合,并将混合物加热至80℃~260℃的温度(优选为180℃~210℃)以进行Diels-Alder反应,从而将马来酸或马来酸酐加成至松香所包含的含共轭双键的酸。
反应后,马来酸改性松香可原样使用,也可以通过如蒸馏等工序进一步精制而后使用。
此外,以同样的方式合成衣康酸改性松香和柠康酸改性松香至上述改性度,并计算和确定改性度。
相对于全部多元羧酸成分的量,该羧酸改性松香的含量比例优选为5重量%~70重量%(优选为10重量%~60重量%)。
其他多元羧酸的优选实例包括二羧酸。二羧酸的实例包括选自由下述物质组成的组中的至少一种:芳香族二羧酸和脂肪族二羧酸,例如,如邻苯二甲酸、间苯二甲酸、对苯二甲酸、1,4-萘二羧酸和2,6-萘二羧酸等芳香族二羧酸;如草酸、丙二酸、马来酸、富马酸、柠康酸、衣康酸、戊烯二酸、琥珀酸、己二酸、癸二酸、壬二酸、二聚酸、具有1~10个碳原子的支化烷基琥珀酸、和含有具有1~20个碳原子的烯基的支化烯基琥珀酸等脂肪族二羧酸;以及这些酸的酸酐和这些酸的烷基(具有1~3个碳原子)酯。其中,从色调剂的耐久性、定影性以及着色剂分散性的角度考虑,优选的是芳香族二羧酸。
其他多元羧酸的实例包括三元以上的多元羧酸。三元以上的多元羧酸的实例包括偏苯三酸、苯均四酸和柠檬酸。
多元醇成分
具有松香的多元醇的优选实例包括含有松香的二醇(下文某些情况中称之为松香二醇)。
本文中,松香二醇是在一个分子中具有两个松香酯基的含松香二醇化合物。并且,松香酯基是指通过由松香中的羧基除去氢原子而形成的残基。
松香二醇可以通过已知方法合成,并且可以例如通过松香与双官能环氧化合物的反应来合成。
此外,作为合成中所用的松香,可以采用与聚酯树脂的缩聚成分所用的松香相同的那些松香。
松香与双官能环氧化合物的反应通常通过松香中羧基与双官能环氧化合物中的环氧基的开环反应来进行。此处,反应温度优选为不低于两种成分的熔点或者为实现均匀混合的温度,具体而言,所述温度通常为60℃~200℃。在反应期间,可以添加促进环氧基的开环反应的催化剂。
松香与双官能环氧化合物的反应所用的催化剂的实例包括如乙二胺、三甲胺和2-甲基咪唑等胺类;如三乙基溴化铵、三乙基氯化铵和丁基三甲基氯化铵等季铵盐类;以及三苯基膦。
松香与双官能环氧化合物的反应通过各种方法中的任一种进行,反应进程通常如下跟踪:在分批式的情况中,将松香和双官能环氧化合物以所需比例放入装配有冷凝器、搅拌器、惰性气体入口和温度计等的能加热的烧瓶中,将混合物加热熔融,并对反应产物取样。反应进展的程度通常通过酸值下降来确定,当反应达到化学计量反应的终点或接近终点时反应结束。
松香和双官能环氧化合物之间的反应比例没有特别限制,但松香与双官能环氧化合物的摩尔比优选为:相对于1摩尔的双官能环氧化合物,松香为1.5摩尔~2.5摩尔。
双官能环氧化合物是一个分子中含两个环氧基的双官能环氧化合物。双官能芳香族环氧化合物的实例包括芳香族二醇的二缩水甘油醚和芳香族二羧酸的二缩水甘油醚;双官能脂肪族环氧化合物的实例包括链式脂肪族二醇的二缩水甘油醚、脂环族二醇的二缩水甘油醚和脂环族环氧化物。
芳香族二醇的二缩水甘油醚的典型实例包括:双酚A、如双酚A的聚氧化烯加合物等双酚A的衍生物、双酚F、如双酚F的聚氧化烯加合物等双酚F的衍生物、双酚S、如双酚S的聚氧化烯加合物等双酚S的衍生物、间苯二酚、叔丁基邻苯二酚和联苯二酚。
芳香族二羧酸的二缩水甘油醚的典型实例包括对苯二甲酸、间苯二甲酸和邻苯二甲酸。
链式脂肪族二醇的二缩水甘油醚的典型实例包括乙二醇、1,2-丙二醇、1,3-丙二醇、1,4-丁二醇、1,5-戊二醇、1,6-己二醇、新戊二醇、1,9-壬二醇、二乙二醇、三乙二醇、聚乙二醇、聚丙二醇和聚丁二醇。
脂环族二醇的二缩水甘油醚的典型实例包括氢化双酚A、如氢化双酚A的聚氧化烯加合物等氢化双酚A的衍生物、和环己烷二甲醇。
脂环族环氧化物的典型实例包括二氧化萜二烯。
双官能环氧化合物可以例如通过二醇与表卤代醇的反应获得,并且可以通过根据数量比进行缩聚反应而具有高分子量。
下面,松香二醇的示例性化合物如下所示,但本示例性实施方式不限于此。
并且,在松香二醇的示例性化合物中,n表示1以上的整数。
基于全部醇成分,松香二醇(具有松香的多元醇)的含量比例为15重量%~98重量%(优选为20重量%~95重量%)。
其他多元醇的实例包括选自由脂肪族二醇和醚化二酚组成的组中的至少一种物质。
脂肪族二醇的实例包括乙二醇、1,2-丙二醇、1,3-丙二醇、1,2-丁二醇、1,3-丁二醇、1,4-丁二醇、2,3-丁二醇、1,4-丁烯二醇、2-甲基-1,3-丙二醇、1,5-戊二醇、新戊二醇、2-乙基-2-甲基丙烷-1,3-二醇、2-丁基-2-乙基丙烷-1,3-二醇、1,6-己二醇、3-甲基-1,5-戊二醇、2-乙基-1,3-己二醇、2,4-二甲基-1,5-戊二醇、2,2,4-三甲基-1,3-戊二醇、1,7-庚二醇、1,8-辛二醇、1,9-壬二醇、1,10-癸二醇、3-羟基-2,2-二甲基丙基-3-羟基-2,2-二甲基丙酸酯、二乙二醇、三乙二醇、聚乙二醇、二丙二醇和聚丙二醇。这些脂肪族二醇可以单独使用或者其中两种以上组合使用。
醚化二酚包括通过使双酚A与氧化烯进行加成反应所获得的二醇,氧化烯的实例包括氧化乙烯和氧化丙烯,相对于1摩尔双酚A,所加成的氧化烯的平均摩尔数优选为2摩尔~16摩尔。
制备特定聚酯树脂的方法的实例包括其中多元羧酸和多元醇用作原料的已知制备方法。作为反应方法,可以应用酯交换反应或者直接酯化反应。并且,可以通过采用在加压下于较高温度进行反应的方法、在减压下进行反应的方法、或者在常压下于惰性气体流中进行反应的方法来加速缩聚反应。在上述反应中,可以使用如选自锑、钛、锡、锌、铝和锰的化合物的至少一种金属化合物等公知的反应催化剂,从而加速反应。相对于100重量份的多元羧酸和多元醇的总量,反应催化剂的添加量优选为0.01重量份~1.5重量份,更优选为0.05重量份~1.0重量份。反应例如在180℃~300℃的温度进行。
此外,当特定聚酯树脂水解时,其分解为单体(多元羧酸成分和多元醇成分)。特定聚酯树脂是,例如,多元羧酸(例如二羧酸)和多元醇(例如二醇)的1:1缩合物,因此,可从分解产物推定树脂的构成。
(特定聚酯树脂的特性)
从色调剂的耐久性和抗沾污性考虑,特定聚酯树脂的重均分子量优选为4000~1000000,更优选为7000~300000。
此外,以下述方式测量特定聚酯树脂的分子量。
使用为“HLC-8120GPC,SC-8020”(由Tosoh Corp.制造,6.0mm ID×15cm)的两个柱,THF(四氢呋喃)用作洗脱剂。关于实验条件,采用下述条件进行测量:样品浓度0.5%,流速0.6ml/分钟,样品注入量10μl,测量温度40℃,RI检测器。并且,采用Tosoh Corp.制造的“聚苯乙烯标准样品TSK Standard”的10个样品:“A-500”、“F-1”、“F-10”、“F-80”、“F-380”、“A-2500”、“F-4”、“F-40”、“F-128”和“F-700”准备校准曲线。
并且,从色调剂的定影性、保存性和耐久性考虑,特定聚酯树脂的软化温度优选为80℃~160℃,更优选为90℃~150℃。
顺便说,软化温度被确定为在升温式(elevated)流动测试仪CFT-500(由ShimadzuCo.制造)中,在模具微孔直径0.5mm、施加负载0.98MPa(10kg/cm2)、加热速率1℃/分钟的条件下,当1cm3样品熔融并使之流动时,流动开始温度和终止温度之间的中心值所对应的温度。
从定影性、保存性和耐久性考虑,特定聚酯树脂的玻璃化转变温度优选为35℃~80℃,更优选为40℃~70℃。软化温度和玻璃化转变温度可通过调整原料单体的组成、聚合引发剂、分子量或催化剂的量等或者通过选择反应条件而容易地调整。
此外,通过采用“DSC-20”(由Seiko Industries and Electronics Co.制造)以恒定加热速度(10℃/分钟)加热10mg样品来测量玻璃化转变温度。
从色调剂充电性的角度考虑,特定聚酯树脂的酸值优选为1mg KOH/g~50mgKOH/g,更优选为3mg KOH/g~30mg KOH/g。
此外,根据JIS K0070采用中和滴定法测量酸值。即,准备适量的样品,添加100ml溶剂(乙醚/乙醇混合液)和几滴试剂(酚酞溶液),在水浴中进行充分的搅拌和混合直至样品溶解。以0.1mol/l氢氧化钾/乙醇溶液对其进行滴定,当试剂的淡红色保持30秒时达到终点。在将酸值表示为A,样品量表示为S(g),滴定中所用的0.1mol/l氢氧化钾/乙醇溶液的量表示为B(ml)并且将f作为0.1mol/l氢氧化钾/乙醇溶液的系数时,计算A=(B×f×5.611)/S。
特定聚酯树脂可以是改性聚酯树脂。改性聚酯的实例包括根据JP-A-11-133668、JP-A-10-239903或JP-A-08-20636等中所描述的方法以苯酚、氨基甲酸酯或环氧类等进行接枝或嵌段的聚酯。
-其他非晶性树脂-
除特定聚酯树脂以外的其他非晶性树脂的实例包括已知粘合剂树脂,例如,如苯乙烯-丙烯酸类树脂等乙烯基类树脂,以及如环氧树脂、聚碳酸酯和聚氨酯等其他树脂。
-结晶性树脂-
结晶性树脂的实例包括结晶性聚酯树脂、聚烷撑树脂和长链烷基(甲基)丙烯酸酯树脂,但从进一步表现因加热所致的粘度剧烈变化并同时满足机械强度和低温定影性的角度考虑,优选的是结晶性聚酯树脂。
作为结晶性聚酯树脂,例如,从实现低温定影性的角度考虑,优选的是由脂肪族二羧酸(包括酸酐及其酰氯)和脂肪族二醇获得的缩聚物。
相对于100重量份的全部粘合剂树脂,结晶性树脂的含量优选为1重量份~20重量份,更优选为5重量份~15重量份。
此外,在本示例性实施方式中,低温定影是指在等于或低于约120℃的温度加热色调剂的情况下定影。
-着色剂-
着色剂可以是例如染料或颜料,但从耐光性和耐水性的角度考虑,优选的是颜料。
作为着色剂,可以采用已知的颜料,例如:炭黑、苯胺黑、苯胺蓝、charcoyl蓝、铬黄、群青蓝、杜邦油红、喹啉黄、亚甲基蓝氯化物、酞菁蓝、孔雀石绿草酸盐、灯黑、玫瑰红、喹吖啶酮、联苯胺黄、C.I.颜料红48:1、C.I.颜料红57:1、C.I.颜料红122、C.I.颜料红185、C.I.颜料红238、C.I.颜料黄12、C.I.颜料黄17、C.I.颜料黄180、C.I.颜料黄97、C.I.颜料黄74、C.I.颜料蓝15:1和C.I.颜料蓝15:3。
作为着色剂,如果需要,可以使用表面经处理的着色剂或颜料分散剂。
通过选择着色剂的类型,可以获得黄色色调剂、品红色色调剂、青色色调剂或黑色色调剂等。
相对于100重量份的粘合剂树脂,着色剂的含量优选为1重量份~30重量份。
-防粘剂-
防粘剂的具体实例包括:如低分子量聚丙烯和低分子量聚乙烯等石蜡;硅树脂;松香;米糠蜡;和巴西棕榈蜡。防粘剂的熔融温度优选为50℃~100℃,更优选为60℃~95℃。
相对于100重量份的粘合剂树脂,防粘剂的含量优选为0.5重量份~15重量份,更优选为1.0重量份~12重量份。
当防粘剂的含量等于或大于0.5重量%时,避免了特别是在无油定影中剥离不良的发生。当防粘剂的含量等于或小于15重量%时,色调剂的流动性不会劣化,并改善了图像形成中的图像品质和可靠性。
-其他添加剂-
作为电荷控制剂,可以使用已知的电荷控制剂,例如,可以使用偶氮类金属络合物、水杨酸金属络合物或含极性基团的树脂类电荷控制剂。
-色调剂颗粒的特性-
色调剂颗粒可以是具有单层结构的色调剂颗粒,或者是由核(芯颗粒)和涂覆在核上的涂覆层(壳层)构成的具有核/壳结构的色调剂颗粒。
优选的是,具有核/壳结构的色调剂颗粒由例如包含粘合剂树脂(特定聚酯树脂和结晶性聚酯树脂)和如果需要的如着色剂和防粘剂等其他添加剂的核和包含粘合剂树脂(本示例性实施方式的聚酯树脂)的涂覆层构成。
色调剂颗粒的体积平均粒径优选为,例如,2.0μm~10μm,更优选为3.5μm~7.0μm。
此外,关于测量色调剂颗粒的体积平均粒径的方法,将0.5mg~50mg测量用样品添加至2ml作为分散剂的表面活性剂(优选为5重量%的烷基苯磺酸钠水溶液)中。将混合物添加至100ml~150ml的电解液中。以超声波分散器将其中悬浮有测量样品的电解液进行分散处理1分钟,通过COULTER MULTISIZER Type II(由Beckmann Coulter Inc.制造)采用孔径为100μm的孔确定粒径为2.0μm~60μm的颗粒的粒径分布。所测量的颗粒数是50,000。
将所获得的粒径分布划分为多个粒径范围(区段),并从较小粒径侧起绘出体积累积分布。将累积分布中的50%所对应的粒径确定为体积平均粒径D50v。
色调剂颗粒的形状系数SF1优选为110~150,更优选为120~140。
本文中,通过下式(1)确定形状系数SF1:
SF1=(ML2/A)×(π/4)×100 式(1)
式(1)中,ML表示色调剂的绝对最大长度,A表示色调剂的投影面积。
并且,SF1值通常可以通过下述方法获得:例如,如下所述,通过图像分析仪对由显微镜或扫描电子显微镜(SEM)捕集的图像进行分析,并计算为数值。即:SF1值可以如下获得:经摄像机将散布在载玻片表面上的颗粒的光学显微图像输入LUZEX图像分析仪中,确定100个颗粒的最大长度和投影面积,通过上式(1)计算所述值,然后取平均值。
(外添剂)
外添剂的实例包括无机颗粒,无机颗粒的实例包括SiO2、TiO2、Al2O3、CuO、ZnO、SnO2、CeO2、Fe2O3、MgO、BaO、CaO、K2O、Na2O、ZrO2、CaO·SiO2、K2O·(TiO2)n、Al2O3·2SiO2、CaCO3、MgCO3、BaSO4和MgSO4。
作为外添剂的无机颗粒的表面可以预先进行疏水处理。疏水处理通过例如将无机颗粒浸渍在疏水处理剂中而进行。疏水处理剂没有特别限制,但其实例包括硅烷类偶联剂、硅油、钛酸酯类偶联剂和铝类偶联剂。这些疏水处理剂可以单独使用或者其两种以上组合使用。
相对于100重量份的无机颗粒,疏水处理剂的量通常为例如1重量份~10重量份。
外添剂的实例包括树脂颗粒(聚苯乙烯、PMMA或三聚氰胺树脂等的树脂颗粒)和清洁活化剂(例如,高级脂肪酸的金属盐,通常以硬脂酸锌为代表,或者氟类聚合物的颗粒粉末)。
相对于100重量份的色调剂颗粒,外添剂的外部添加量例如优选为0.01重量份~5重量份,更优选为0.01重量份~2.0重量份。
外添剂的一次粒径例如优选为5nm~2μm,更优选为5nm~500nm。
(制备色调剂的方法)
下文中将描述制备色调剂的方法。
首先,可以通过干式制备法(例如,混炼粉碎法)和湿式制备法(例如,凝集聚并法、悬浮聚合法、溶解悬浮造粒法、溶解悬浮法和溶解乳化凝集聚并法)中的任意一种方法来制备含特定聚酯树脂的色调剂颗粒。制备方法没有特别限制,可以选择使用公知的制备方法中的任意一种。
这些方法中,优选的是通过凝集聚并法获得色调剂颗粒。
另外,色调剂通过例如将外添剂添加至由此获得的干燥色调剂颗粒中并将其混合而制备。混合优选使用例如V型混合器、亨舍尔混合器或Loedige混合器等进行。此外,如果需要,可以使用振动筛或风力筛等除去色调剂中的粗颗粒。
<载体>
载体具有芯材颗粒和涂覆芯材颗粒表面的涂覆树脂层。
下面提供载体的细节。
(芯材颗粒)
作为芯材颗粒,可以使用用作载体用芯材颗粒的任何已知颗粒,但其实例包括磁性颗粒,具体而言,磁性金属颗粒(例如铁、钢、镍或钴等的颗粒)或磁性氧化物颗粒(例如铁氧体或磁铁矿等的颗粒)。例如,所述颗粒可以是其中颗粒分散在树脂中的磁性颗粒分散型树脂颗粒。
(涂覆树脂层)
涂覆树脂层的硬度为色调剂颗粒的硬度的1.2倍~2.0倍。
涂覆树脂层用树脂可以是满足上述硬度的任何树脂,其实例包括聚乙烯、聚丙烯、聚苯乙烯、聚乙酸乙烯酯、聚乙烯醇、聚乙烯醇缩丁醛、聚氯乙烯、聚乙烯醚、聚乙烯酮、氯乙烯-乙酸乙烯酯共聚物、苯乙烯-丙烯酸共聚物、具有有机硅氧烷键的纯硅酮树脂及其改性产物、氟树脂、聚酯、聚碳酸酯、酚醛树脂、环氧树脂和丙烯酸类树脂。
本文中,满足上述硬度的涂覆树脂层用树脂的优选实例包括:包含选自(甲基)丙烯酸二环戊基酯、(甲基)丙烯酸异冰片基酯、(甲基)丙烯酸金刚烷基酯、(甲基)丙烯酸降冰片基酯和(甲基)丙烯酸羟基萘基酯的至少一种物质作为聚合成分的树脂,更优选的是,包含选自(甲基)丙烯酸二环戊基酯和(甲基)丙烯酸异冰片基酯的至少一种物质作为聚合成分的树脂。
据认为,当涂覆树脂层具有涂覆树脂层用树脂包含选自上述聚合成分的至少一种作为聚合成分的构成时,其倾向于容易实现上述硬度,并因此容易表现本申请的效果。
据推测,原因是由于上述聚合成分具有其中高分子量基团结合到(甲基)丙烯酸的酯基的结构,当由聚合形成树脂时,容易实现所述硬度。
此外,据认为,当涂覆树脂层具有其中涂覆树脂层用树脂包含选自上述聚合成分的至少一种作为聚合成分的构成时,可抑制剥落。
作为涂覆树脂层用树脂的聚合成分,除了上述聚合成分外,还可以包含其他聚合成分,以调节涂覆树脂层的硬度。
其他聚合成分的具体实例包括(甲基)丙烯酸、(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸丙酯、苯乙烯和(甲基)丙烯酸二乙基氨基乙基酯。
从实现涂覆树脂层的硬度并因而抑制涂覆树脂层的剥落的角度考虑,相对于涂覆树脂层用树脂中的全部聚合成分,作为涂覆树脂层用树脂的聚合成分的(甲基)丙烯酸二环戊基酯、(甲基)丙烯酸异冰片基酯、(甲基)丙烯酸金刚烷基酯、(甲基)丙烯酸降冰片基酯或(甲基)丙烯酸羟基萘基酯的含量优选为30重量%~95重量%,更优选为50重量%~85重量%,进而更优选为60重量%~80重量%。
载体的涂覆树脂层的逸出功优选为等于或小于4.55eV。
据认为,当涂覆树脂层的逸出功优选为等于或小于4.55eV时,其倾向于变得比色调剂颗粒中所含的特定聚酯树脂的逸出功低,因此,改善了向色调剂提供电荷的能力。
涂覆树脂层的逸出功优选为4.2eV~4.5eV,更优选为4.3eV~4.4eV。
涂覆树脂层的逸出功如下计算。
将涂覆树脂溶解在四氢呋喃中从而获得2重量%的四氢呋喃溶液。将该溶液涂布在镀金铜基板上并使四氢呋喃蒸发以获得厚度为约0.5μm的薄膜。而后,在120℃的烘箱中将溶液干燥5小时以获得测量用样品。在温度为23℃、湿度为50%的环境下,采用Kelvin探针在接触电位差测量装置中获得参比电极与涂覆有涂覆树脂的样品基板之间的接触电位差。参比电极的逸出功采用光电子发射装置(由Riken Keiki Co.Ltd.制造的AC2)测量,从参比电极的逸出功中减去接触电位差得出作为涂覆树脂的逸出功的值。当涂覆树脂的逸出功小于参比电极的逸出功时,接触电位差为正,否则为负。另外,关于此细节,参考JOHOKIKO Co.Ltd.的“Case Studies ofMeasurement/Evaluation and Control/Use of Work Function/Ionization Potential(2010)”。
另外,色调剂颗粒中所含的特定聚酯树脂的逸出功以相同方式计算。
例如,如导电性材料等其他添加剂也可以包含在涂覆树脂层中。
导电性颗粒的实例包括:炭黑;各种金属粉末;如二氧化钛、氧化锡、磁铁矿和铁氧体等金属氧化物。这些物质可以单独使用,或者其两种以上组合使用。其中,就良好的制备稳定性、成本、导电性等而言,优选的是炭黑颗粒。炭黑的种类没有特别限制,但就优异的制备稳定性而言,优选的是DBP吸油量为约50ml/100g~250ml/100g的炭黑。
为了在芯材颗粒的表面上形成涂覆树脂层,例如,可以举出下述方法:将涂覆树脂和如果需要的各种添加剂溶解在适当的溶剂中,并将由此形成的涂覆树脂层形成用溶液进行涂覆。溶剂没有特别限制,可以考虑所用的涂覆树脂的种类和/或涂覆适用性来适当地选择。
所述方法的具体实例包括:其中将芯材颗粒浸渍在涂覆树脂层形成用溶液中的浸渍法,其中将涂覆层形成用溶液喷雾在芯材颗粒表面上的喷雾法,其中在芯材颗粒通过流态化空气漂浮的同时喷雾涂覆树脂层形成用溶液的流化床法,和其中将载体的芯材颗粒与涂覆树脂层形成用溶液在混炼涂布机中混合并除去溶剂的混炼涂布机法。
此处,相对于载体的总重量,涂覆在芯材颗粒上的涂覆树脂层的量例如优选为0.5重量%以上(更优选为0.7重量%~6重量%,进而更优选为1.0重量%~5.0重量%)。
该涂覆量如下确定。
在涂覆树脂可溶于溶剂的情况中,将精确称量的载体溶解在可溶溶剂(例如,甲苯)中,以磁铁保持住磁性粉末,通过清洗除去其中溶解有涂覆树脂的溶液。将该操作重复数次,留下已除去涂覆树脂的磁性粉末。称量干燥后的磁性粉末的重量,然后将差值除以载体的量以计算涂覆量。
具体而言,称量20.0g载体并置于烧杯中,向其中添加100g甲苯,并以搅拌叶片搅拌混合物10分钟。将磁铁放置在烧杯底部之下,然后以芯材(磁性粉末)不会流出的方式用甲苯清洗。将该操作重复四次,并将清洗后的烧杯干燥。干燥后,测量磁性粉末的量并采用公式[(载体的量-清洗后的磁性粉末的量)/载体的量]来计算涂覆量。
另一方面,在涂覆树脂不溶于溶剂的情况中,采用Thermo plus EVO II差示热重分析仪TG8120(由Rigaku Corporation制造)在氮气氛围中于室温(25℃)~1000℃的温度加热载体,并由载体重量的减少来计算涂覆量。
(制备显影剂的方法)
本示例性实施方式的静电荷图像显影剂是通过混合色调剂和载体而形成的双成分显影剂。
在双成分显影剂中色调剂和载体的混合比(重量比)为:色调剂:载体的比为约1:100~30:100,更优选为约3:100~20:100。
[图像形成装置/图像形成方法]
接下来,将描述本示例性实施方式的图像形成装置/图像形成方法。
本示例性实施方式的图像形成装置具有:图像保持体;对图像保持体的表面进行充电的充电单元;在经充电的图像保持体的表面上形成静电荷图像的静电荷图像形成单元;容纳有静电荷图像显影剂,并通过静电荷图像显影剂使图像保持体上形成的静电荷图像显影,从而形成色调剂图像的显影单元;将图像保持体上形成的色调剂图像转印到记录介质上的转印单元;和将转印到记录介质上的色调剂图像定影的定影单元。此外,采用本示例性实施方式的静电荷图像显影剂作为所述静电荷图像显影剂。
此外,在本示例性实施方式的图像形成装置中,例如,包含显影单元的部分可以是能在图像形成装置上安装和拆卸的盒结构(处理盒),并且优选使用容纳有本示例性实施方式的静电荷图像显影剂并包含显影单元的处理盒作为所述处理盒。
本示例性实施方式的图像形成方法包括:对图像保持体的表面进行充电的步骤;在经充电的图像保持体的表面上形成静电荷图像的步骤;保持静电荷图像显影剂并通过静电荷图像显影剂使图像保持体上形成的图像显影,从而形成色调剂图像的步骤;将图像保持体上形成的色调剂图像转印到记录介质上的步骤;和将转印到记录介质上的色调剂图像定影的步骤。此外,采用本示例性实施方式的静电荷图像显影剂作为所述静电荷图像显影剂。
下面将描述本示例性实施方式的图像形成装置的实例,但本发明不限于此。另外,描述了附图中所示的主要部分,省略对其他部分的描述。
图1是显示四串联式彩色图像形成装置的示意性构造图。图1中所示的图像形成装置具有第一至第四电子照相图像形成单元10Y、10M、10C和10K(图像形成单元),它们根据分色图像数据分别输出黄色(Y)、品红色(M)、青色(C)和黑色(K)的图像。这些图像形成单元(下文某些情况中简称为“单元”)10Y、10M、10C和10K在水平方向上以预定间隔并排排列。并且,这些单元10Y、10M、10C和10K可以是能在图像形成装置的主体上安装或拆卸的处理盒。
在图中的各单元10Y、10M、10C和10K的上方,连接有作为中间转印介质的中间转印带20经过各单元。中间转印带20卷绕在沿图中从左到右的方向彼此分开设置的驱动辊22和与中间传送带20的内表面接触的支撑辊24上,从而沿从第一单元10Y到第四单元10K的方向运转。并且,使用弹簧等(未示出)对支撑辊24在与驱动辊22分离的方向上施加力,并对卷绕在这两个辊上的中间转印带20施加张力。在中间转印带20的图像保持体侧,设置有与驱动辊22相对的中间转印介质清洁装置30。
并且,将色调剂盒8Y、8M、8C和8K中储存的黄色、品红色、青色和黑色这四色色调剂分别供给至相应的各单元10Y、10M、10C和10K的显影装置(显影单元)4Y、4M、4C和4K。
由于上述第一至第四单元10Y、10M、10C和10K具有相同的构成,因此仅将设置于中间转印带运转方向的上游的形成黄色图像的第一单元10Y作为代表性实例进行说明。对与第一单元10Y相同的部件指定品红色(M)、青色(C)和黑色(K)代替黄色(Y),由此省略对第二至第四单元10M、10C和10K的说明。
第一单元10Y具有起图像保持体作用的感光体1Y。感光体1Y周围依次设置有:充电辊2Y,它可将感光体1Y的表面充电到预定电位;曝光装置(静电潜像形成单元)3,它可根据分色图像信号以激光束3Y对经充电的表面进行曝光,以形成静电荷图像;显影装置(显影单元)4Y,它可以向静电潜像供应带电的色调剂从而使静电潜像显影;一次转印辊(一次转印单元)5Y,它将经显影的色调剂图像转印到中间转印带20上;和感光体清洁装置(清洁单元)6Y,它可以除去一次转印后残留在感光体1Y表面上的色调剂。
另外,一次转印辊5Y设置于中间转印带20的内侧,并且设置在与感光体1Y相对的位置上。此外,一次转印辊5Y、5M、5C和5K各自与施加一次转印偏压的偏压电源(未示出)相连。在控制单元(未示出)的控制下,各偏压电源可以改变施加在各一次转印辊上的转印偏压。
下面描述在第一单元10Y中形成黄色图像的操作。首先,在操作前,通过充电辊2Y将感光体1Y的表面充电至约-600V至约-800V的电位。
通过在导电性(20℃时体积电阻率等于或小于1×10-6Ωcm)基体上层积设置感光层,形成感光体1Y。感光层通常具有高电阻(与一般树脂的电阻相同或相近)。当以激光束3Y照射感光层时,激光束照射部分的比电阻发生改变。然后,根据控制单元(未示出)发送的黄色图像数据,激光束3Y经由曝光装置3发射到经充电的感光体1Y的表面上。激光束3Y发射到感光体1Y上的感光层,由此在感光体1Y的表面上形成具有黄色印刷图案的静电荷图像。
静电荷图像是通过充电在感光体1Y表面上形成的图像,是通过下述方式形成的所谓的负潜像:通过激光束3Y,感光层的照射部分的比电阻降低,带电电荷在感光体1Y的表面上流动,相反,未以激光束3Y照射的部分的电荷则保持不变。
感光体1Y上由此形成的静电荷图像随着感光体1Y的运行而旋转到预定显影位置。然后,通过显影装置4Y使感光体1Y上的静电荷图像在显影位置处形成为可视图像(显影图像)。
在显影装置4Y中,容纳有例如至少包含黄色色调剂和载体的本示例性实施方式的静电荷图像显影剂。黄色色调剂在显影装置4Y中搅拌,从而使色调剂摩擦带电,在具有与感光体1Y上的电荷相同极性(负极性)的电荷的同时,并保持在显影辊(显影剂保持体)上。当感光体1Y的表面穿过显影装置4Y时,黄色色调剂静电附着在感光体1Y表面上的经除电的潜像部分上,由此用黄色色调剂使潜像显影。其上形成有黄色色调剂图像的感光体1Y以预定速率连续地运行,并且将感光体1Y上已显影的色调剂图像传送到一次转印位置。
当将感光体1Y上的黄色色调剂图像传送至一次转印处时,对一次转印辊5Y施加一次转印偏压,使从感光体1Y指向一次转印辊5Y方向的静电力作用于色调剂图像,从而将感光体1Y上的色调剂图像转印到中间转印带20上。此时施加的转印偏压具有与色调剂的极性(-)相反的极性(+),并且在第一单元10Y中通过控制单元(未示出)将转印偏压控制在例如约+10μA。
另一方面,通过清洁装置6Y除去并收集感光体1Y上残留的色调剂。
另外,根据第一单元,也对施加于第二单元10M以后的一次转印辊5M、5C和5K的一次转印偏压进行控制。
这样,使已通过第一单元10Y转印有黄色色调剂图像的中间转印带20依次传送通过第二至第四单元10M、10C和10K,使各种颜色的色调剂图像与黄色色调剂图像重叠,实现多重转印。
通过第一至第四单元而多重转印有四种颜色的色调剂图像的中间转印带20到达二次转印部,所述二次转印部由中间转印带20、与中间转印带20的内表面相接触的支撑辊24和设置在中间转印带20的图像保持面侧的二次转印辊(二次转印单元)26形成。通过供给机构以预定时机向二次转印辊26和中间转印带20相互压接的部位供应记录纸(记录介质)P,并向支撑辊24施加二次转印偏压。此时所施加的转印偏压具有与色调剂极性(-)相同的极性(-),从中间转印带20指向记录纸P的方向的静电力作用于色调剂图像,由此将中间转印带20上的色调剂图像转印到记录纸P上。根据由用于检测二次转印部的电阻的电阻检测单元(未示出)所检测的电阻来确定这种情况下的二次转印偏压,并控制电压。
随后,将记录纸P传送至定影设备(辊状定影单元)28中的一对定影辊的压接部(咬合部),将色调剂图像定影到记录纸P上从而形成定影图像。
其上转印色调剂图像的记录介质的实例包括电子照相方式的复印机或打印机中所使用的普通纸或OHP片。
为了进一步提高定影后的图像表面的光滑度,还优选光滑的记录介质表面,例如,优选使用通过在普通纸张表面涂覆树脂等而形成的涂布纸或用于印刷的铜版纸等。
在完成彩色图像在记录纸P上的定影后,将记录纸P排出到排出部,完成一系列彩色图像形成操作。
在作为实例的上述图像形成装置中,色调剂图像经由中间转印带20转印到记录纸P上。然而,图像形成装置的构造并不限于此,也可将色调剂图像从感光体直接转印到记录纸上。
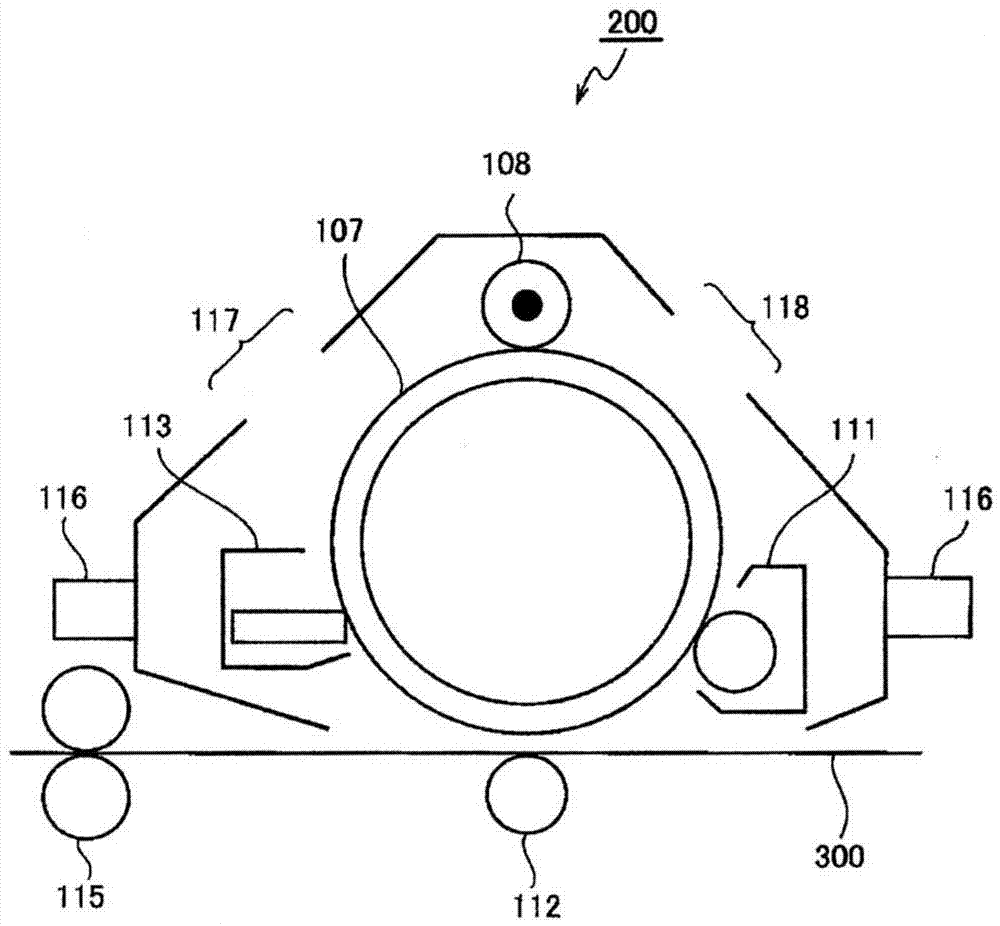
(处理盒和色调剂盒)
图2是显示容纳有本示例性实施方式的静电荷图像显影剂的处理盒的实例的优选示例性实施方式的示意性构造图。处理盒200包含用安装轨道116组合并一体化的感光体107、充电辊108、显影设备111、感光体清洁设备113、曝光用开口118和除电曝光用开口117。图2中的附图标记300表示记录介质。
并且,处理盒200形成为能在包含转印设备112、定影设备115和未示出的其他组件的图像形成装置上安装和拆卸。
图2所示的处理盒200包含充电设备108、显影设备111、清洁设备113、曝光用开口118和除电曝光用开口117,但可以选择性地组合这些设备。本示例性实施方式的处理盒包含感光体107,以及选自由充电设备108、显影设备111、清洁设备(清洁单元)113、曝光用开口118和除电曝光用开口117组成的组中的至少一种。
接下来将描述本示例性实施方式的色调剂盒。本示例性实施方式的色调剂盒是能在图像形成装置上安装或拆卸,并且至少容纳有静电荷图像显影用色调剂(用于分发而供给至图像形成装置中设置的显影单元)的色调剂盒。
另外,图1所示的图像形成装置是具有以可安装或拆卸的方式设置有色调剂盒8Y、8M、8C和8K的结构的图像形成装置。显影设备4Y、4M、4C和4K经由色调剂供应管(未示出)与对应于各颜色所用的各个显影设备的色调剂盒连接。此外,当色调剂盒中容纳的色调剂量变低时,更换色调剂盒。
实施例
下文中,将参照实施例描述本示例性实施方式,但本示例性实施方式并不限于下述实施例。并且,在实施例中,除非另有说明,否则“份”和“%”分别表示“重量份”和“重量%”。
[色调剂]
<羧酸改性松香>
(富马酸改性松香的合成)
将500g妥尔松香和100g富马酸放入装配有分离管和冷凝器的2升烧瓶中,并使混合物在200℃进行反应4小时,从而获得富马酸改性松香(富马酸改性松香酸)。
对于所获得的富马酸改性松香的特性,改性度为85。
(马来酸改性松香的合成)
将500g精制松香和100g马来酸酐添加至2升烧瓶中,并使混合物在220℃进行反应7小时。马来酸改性度为100。
<松香二醇>
(松香二醇(1)的合成)
将113份作为双官能环氧化合物的双酚A二缩水甘油醚(商品名jER828,由Mitsubishi Chemical Industries制造)、200份作为松香成分的已通过蒸馏(蒸馏条件:6.6kPa和220℃)进行了精制处理的脂松香、和0.4份作为反应催化剂的四乙基溴化铵(由Tokyl Kasei Kogyo Co.,Ltd.制造)放入装配有搅拌设备、加热设备、冷凝器和温度计的不锈钢反应容器中,并加热至130℃以进行松香的酸基和环氧化合物的环氧基之间的开环反应。反应在同一温度继续进行4小时,并在酸值达到0.5mg KOH/g时停止反应,从而获得作为示例性化合物提及的松香二醇(1)。
(松香二醇(2)的合成)
将58份作为双官能环氧化合物的乙二醇二缩水甘油醚(商品名EX-810,由NagaseChemtex制造)、200份作为松香成分的歧化松香(商品名Pine Crystal KR614,由Arakawa Chemical Industries,Ltd.制造)、和0.4份作为反应催化剂的四乙基溴化铵(由Tokyl Kasei Kogyo Co.,Ltd.制造)放入装配有搅拌设备、加热设备、冷凝器和温度计的不锈钢反应容器中,并加热至130℃的温度以进行松香的酸基和环氧化合物的环氧基之间的开环反应。反应在同一温度继续进行4小时,并在酸值达到0.5mg KOH/g时停止反应,从而获得作为示例性化合物提及的松香二醇(2)。
<特定聚酯树脂>
-特定聚酯树脂1的合成-
将300份作为醇成分的松香二醇(1)、53份作为多元羧酸成分的对苯二甲酸(由Wako Pure Chemical Industries,Ltd.制造)和0.3份作为反应催化剂的钛酸四正丁酯(由Tokyo Kasei Kogyl Co.,Ltd.制造)放入装配有搅拌设备、加热设备、冷凝器、温度计、分离设备和氮气入口的不锈钢反应容器中,在氮气氛围中搅拌的同时使混合物在230℃进行缩聚反应7小时。经确认,分子量和酸值达到各自的所需值,由此合成出特定聚酯树脂1。
重均分子量、数均分子量、玻璃化转变温度和软化温度的测量结果示于表1中。
-特定聚酯树脂2的合成-
以与特定聚酯树脂1相同的方式合成特定聚酯树脂2,不同之处在于多元羧酸成分和多元醇成分的种类和添加量根据表1改变。
重均分子量、数均分子量、玻璃化转变温度和软化温度的测量结果示于表1中。
表1
-特定聚酯树脂3~5和比较用聚酯树脂1的合成-
以与特定聚酯树脂1相同的方式合成对应于上述1)、3)和4)任一类型的缩聚物的特定聚酯树脂3~5和比较用聚酯树脂1,不同之处在于根据表2改变组成。
重均分子量、数均分子量、玻璃化转变温度和软化温度的测量结果示于表2中。
表2
1)双酚A氧化乙烯2摩尔加合物
2)双酚A氧化丙烯2摩尔加合物
<色调剂颗粒>
(树脂颗粒分散液1的制备)
将100重量份特定聚酯树脂1放入装配有搅拌器的反应器中,溶解并在120℃混合30分钟,然后将中和用水溶液(通过将1.0重量份十二烷基苯磺酸钠和1.0重量份的1N的NaOH水溶液溶解在800重量份已加热至95℃的离子交换水中而形成)放入烧瓶中,以均质器(ULTRA TURRAX,由IKA Co.,Ltd.制造)进行乳化5分钟,然后在超声波浴中振荡10分钟。之后,将烧瓶在室温水(25℃)中冷却,由此获得树脂颗粒分散液1,其中树脂颗粒的中值粒径为250nm,固体含量为20重量%。
(树脂颗粒分散液2~5和比较用树脂颗粒分散液1的制备)
以与树脂颗粒分散液1相同的方式制备树脂颗粒分散液2~5和比较用树脂颗粒分散液1,不同之处在于使用相应的特定聚酯树脂2~5和比较用聚酯树脂1代替特定聚酯树脂1。
(比较用树脂颗粒分散液1的制备)
以与树脂颗粒分散液1相同的方式制备比较用树脂颗粒分散液1,不同之处在于使用相应的比较用聚酯树脂1代替特定聚酯树脂1。
(着色剂颗粒分散液1的制备)
·青色颜料(铜酞菁,C.I.Pigment Blue 15:3,由Dainichiseika Color & ChemicalsMfg.Co.,Ltd.制造):50重量份
·阴离子表面活性剂(NEOGEN R,由Dai-ichi Kogyo Seiyaku Co.,Ltd.制造):5重量份
·离子交换水:200重量份
将上述成分混合溶解,使用均质器(ULTRA TURRAX,由IKA Co.,Ltd.制造)分散5分钟,并在超声波浴中分散10分钟,从而获得中值粒径为190nm、固形物浓度为21.5%的青色着色剂颗粒分散液1。
(防粘剂颗粒分散液1的制备)
·阴离子表面活性剂(NEOGEN R,由Dai-Ichi Kogyo Seiyaku Co.,Ltd.制造):2重量份
·离子交换水:800重量份
·石蜡(HNP-9,由Nippon Seiro Co.,Ltd.制造):200重量份
将上述成分混合、加热至120℃,并用压力排出型Gaulin均质器进行分散处理,从而获得体积平均粒径为170nm的20重量%防粘剂分散液。
(色调剂颗粒1的制备)
·树脂颗粒分散液1:315重量份(树脂63重量份)
·着色剂颗粒分散液1:40重量份(颜料8.6重量份)
·防粘剂颗粒分散液1:40重量份(防粘剂8.0重量份)
·聚氯化铝:0.15重量份
·离子交换水:300重量份
根据上述配方,在圆形不锈钢瓶中用均质器(ULTRA TURRAX T50,由IKA Co.,Ltd.制造)将这些成分混合并分散,然后在搅拌下于加热用油浴中将烧瓶加热至42℃,并在42℃保持60分钟。然后,将105重量份的树脂颗粒分散液1(21重量份树脂)添加至其中,并搅拌混合物。之后,以0.5摩尔/升的氢氧化钠水溶液将体系的pH调节为6.0,然后在搅拌下加热至95℃。在温度升高到95℃时,体系中的pH通常降低到等于或小于5.0,但进一步向其中逐滴添加氢氧化钠水溶液,从而保持pH不会等于或小于5.5。
反应完成后,将混合物冷却、过滤、以离子交换水洗涤,然后通过Nutsche吸滤法进行固液分离。并且,将所得物进一步分散在40℃的3000份离子交换水中,以300rmp搅拌洗涤15分钟。该洗涤操作重复5次,并通过Nutsche吸滤法对所得物进行固液分离。之后,进行12小时真空干燥以获得色调剂颗粒。将所获得的色调剂颗粒1作为色调剂1。
(色调剂颗粒2~5和比较用色调剂颗粒1的制备)
以与色调剂颗粒1相同的方式制备各色调剂颗粒2~5以及比较用色调剂颗粒1,不同之处在于使用相应的树脂颗粒分散液2~5和比较用树脂颗粒分散液1代替树脂颗粒分散液1。
(色调剂1的制备)
将0.5份二氧化硅(商品名:R812,由NipponAerosil Co.,Ltd.制造)添加至色调剂颗粒1(100份)中,并通过高速混合器混合以获得色调剂1。
<色调剂2~5和比较用色调剂1的制备>
以与色调剂1相同的方式制备各色调剂2~5和比较用色调剂1,不同之处在于使用相应的色调剂颗粒2~5以及比较用色调剂颗粒1代替色调剂颗粒1。
[载体]
<涂覆树脂层>
(涂覆树脂层用溶液1)
-树脂的合成-
·甲基丙烯酸二环戊基酯:80重量份
·甲基丙烯酸甲酯:20重量份
·2,2-偶氮二异丁酸二甲酯(商品名V-601,由Wako Pure Chemical Industries,Ltd.制备):2重量份
采用上述组成在下述条件下进行聚合。
将单体和反应引发剂V-601(共102重量份)和400重量份甲苯一起放入容器中,向其中吹入氮气从而在60℃进行反应5小时,由此获得聚合树脂。在温度恢复到室温(25℃)后停止反应,将溶液倒入甲醇中,使树脂再沉淀、过滤并取出。将该树脂在真空下干燥以除去过量的挥发性成分,从而获得所需树脂。
-涂覆树脂层用溶液1的合成-
将1.6重量份如上制备的树脂、下述成分和玻璃珠(粒径:1mm,与甲苯相同量)放入砂磨机(由Kansai Paint Co.,Ltd.制造)以1200rmp的转速处理30分钟,从而制备固体含量为12.6%的涂覆树脂层用溶液1。
·炭黑“VXC72(由Cabot Corporation制造)”:0.12重量份
·甲苯:14重量份
·交联三聚氰胺树脂颗粒(平均粒径0.3μm,不溶于甲苯):0.3重量份
-涂覆树脂层用溶液2~7和涂覆树脂层用比较溶液1的合成-
以与涂覆树脂层用溶液1相同的方式制备涂覆树脂层用溶液2~7和涂覆树脂层用比较溶液1,不同之处在于树脂组成和其他成分的种类和添加量根据表3改变。
<载体的制备>
(载体1)
将2000重量份铁氧体颗粒放入5升真空脱气型混炼机中,并将320重量份涂覆树脂层用溶液1进一步添加至其中。在搅拌混合物的同时,在60℃将压力降至-200mmHg,并将混合物混合15分钟,进行升温/减压,并在94℃/-720mmHg搅拌并干燥30分钟,从而获得涂覆颗粒,其中铁氧体颗粒涂覆有包含涂覆树脂层用溶液1的涂覆树脂层。
接下来,采用75μm网眼筛对所获得的颗粒进行分级从而获得载体1。
(载体2~7和比较用载体1~2)
以与载体1同样的方式制备载体2~7和比较用载体1~2,不同之处在于以根据表3制备的涂覆树脂层用溶液2~7和涂覆树脂层用比较溶液1~2来涂覆铁氧体颗粒。
表3
(实施例1)
(显影剂1)
将7重量份色调剂1添加至100重量份载体1中,并以滚筒振动筛混合器(tumbler-shaker mixer)进行混合从而获得显影剂1。
如下评估所获得的显影剂1。
结果示于表4中。
<评价>
(涂覆树脂层硬度/色调剂颗粒硬度)
通过上述方法,测量涂覆树脂层的硬度和色调剂颗粒的硬度,并计算“涂覆树脂层硬度/色调剂颗粒硬度)。
(色调剂充电量)
采用显影剂1在富士施乐株式会社制造的DocuCentre III 3000中于10000张纸上进行图像形成。具体而言,在28℃/85%RH的环境下进行2%图像面积的印刷。
在进行图像形成之前,以及在10000张纸上进行图像形成之后,从显影装置的显影套筒中收集0.5g显影剂,并通过吹放(blow-off)法由Toshiba Chemical制造的TB200测定充电量。
(外添剂对载体的污染)
收集显影器中磁性套筒上的显影剂,放入装有1重量%非离子表面活性剂水溶液的烧杯中,并进行搅拌。磁铁由烧杯外部接触,并在保持住载体的同时除去其中分散有由载体剥离的色调剂的水溶液。重复这些操作,从显影剂中除去色调剂,并取出载体。再次将载体干燥并通过荧光X射线测量源自外添剂的二氧化硅的Si元素的量,从而评估载体的污染。
评估标准如下。
A:Si的重量%比例小于0.02
B:Si的重量%比例小于0.05且在0.02以上
C:Si的重量%比例小于0.1且在0.05以上
D:Si的重量%比例为0.1以上。
此外,在评估标准中,B表示在可接受范围内的值。
(逸出功)
通过上述方法测量色调剂颗粒中包含的特定聚酯树脂和涂覆树脂层的逸出功。
(实施例2~11和比较例1~2)
以与显影剂1相同的方式制备显影剂2~13,不同之处在于根据表4改变各色调剂和载体的种类,并以与实施例1中相同的方式进行评估。
结果示于表4中。
表4
从上述结果可以看出,在实施例中,与比较例相比,即使在图像形成之后色调剂的充电量也很高,抑制了载体的污染,并且改善了显影剂的使用寿命。
提供对本发明示例性实施方式的前述描述是为了说明和描述的目的。并非试图穷尽或将本发明限制于所披露的确切形式。显然,许多改进和变化对于本领域技术人员是显而易见的。选择并描述所述示例性实施方式是为了能够最好地解释本发明的原理及其实际应用,由此使得本领域的其他技术人员能够理解适用于预计的特定用途的本发明的各种实施方式和各种改进方案。本发明的范围由所附权利要求及其等同形式所限定。
- 还没有人留言评论。精彩留言会获得点赞!